Результат интеллектуальной деятельности: КОРОТКИЙ ПУТЬ СИНТЕЗА 1,6:2,3-ДИАНГИДРО-β-D-МАННОПИРАНОЗЫ
Вид РИД
Изобретение
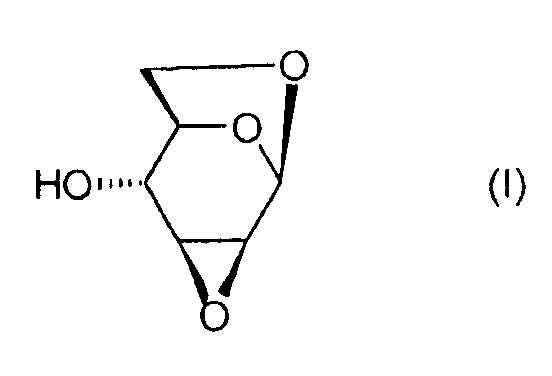
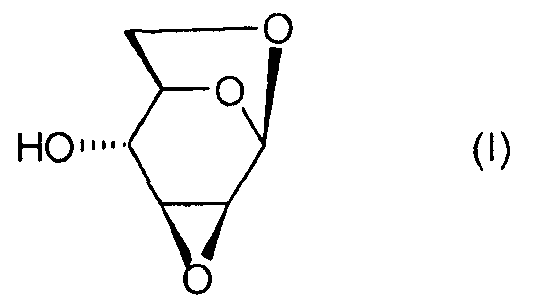
Настоящее изобретение относится к новому способу получения 1,6:2,3-диангидро-β-D-маннопиранозы, называемой далее "эпоксидом Черни" или "соединением (I)" и соответствующей следующей формуле, в которой жирная черта означает связь, находящуюся выше плоскости пиранозного цикла:
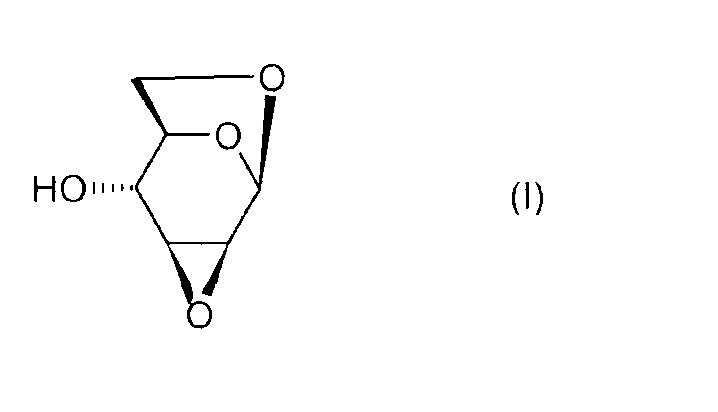
или же, согласно другому представлению:
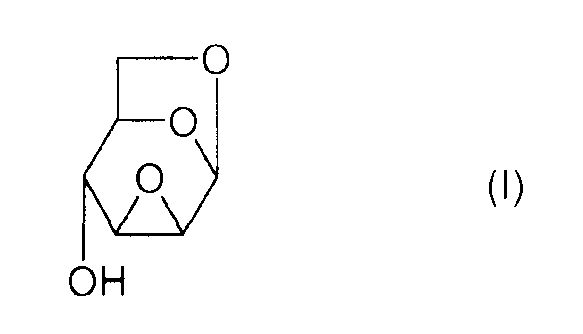
Соединение (I) и, в общем, соединения семейства 1,6:(2,3 и 3,4)-диангидро-β-D-гексопираноз были описаны в основном чешским химиком Милославом Черни. В литературе имеется три способа получения эпоксида Черни (I) из соединения 1 (1,6:3,4-диангидро-4-O-тозил-β-D-галактопираноза):
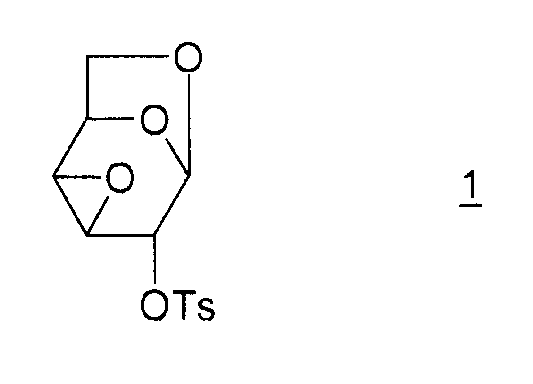
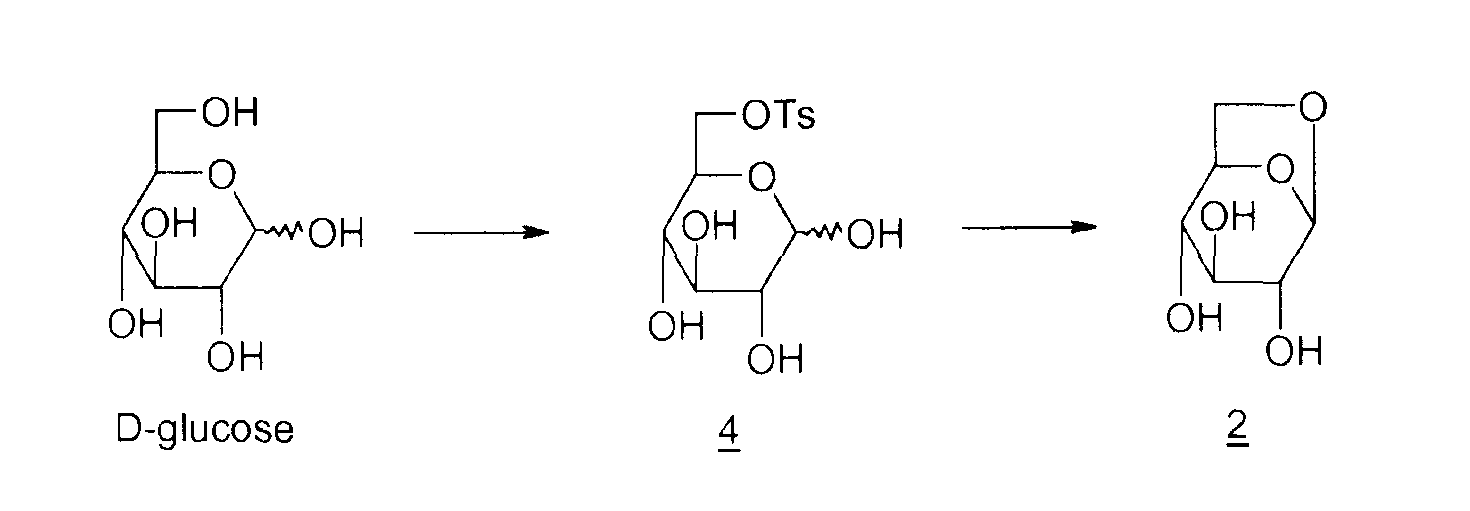
Соединение 1 получают из левоглюкозана 2 (или 1,6-ангидро-β-D-глюкопиранозы), как показано ниже (M. Cerny et al., Collect. Czech. Chem. Commun., 1961, vol. 26, p. 2542-2550):
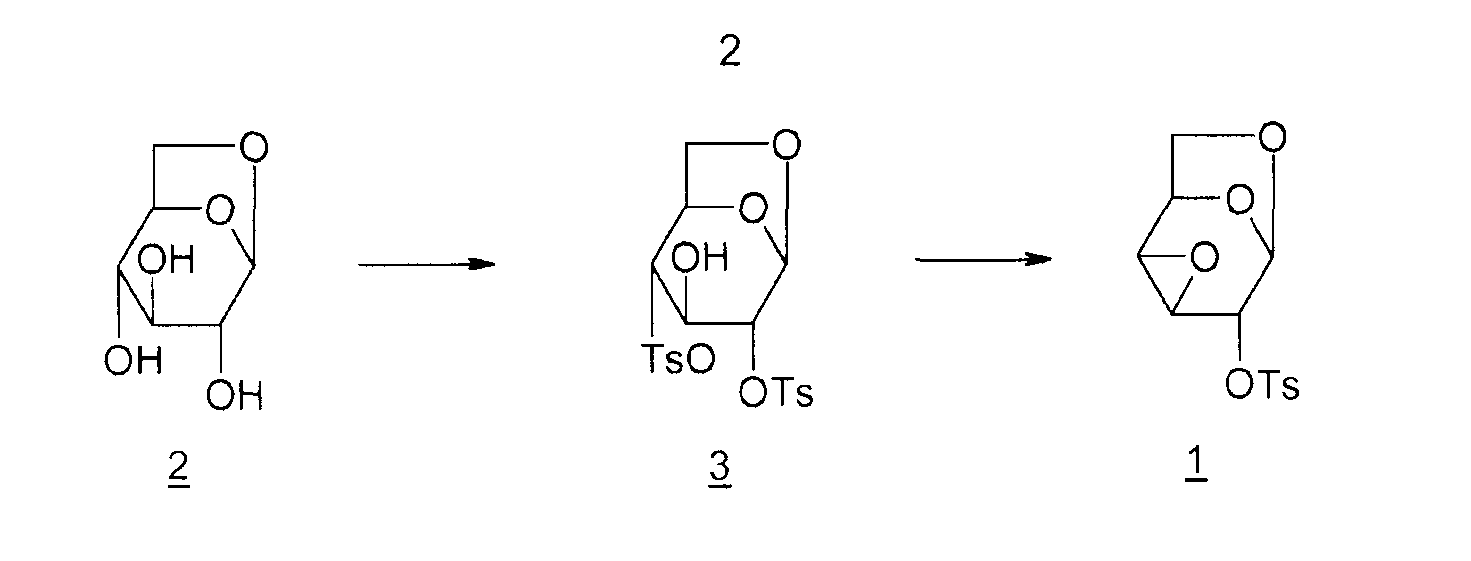
Дитозильное производное 3 (1,6-ангидро-2,4-ди-O-тозил-β-D-глюкопираноза) получают селективно (80%). Остальные 20% состоят в основном из тритозильного производного. Общий выход для превращения соединения 2 в соединение 1 составляет 55%.
Способов получения левоглюкозана 2 много; наиболее распространенными в промышленности, помимо пиролиза крахмала и целлюлозы, описанного еще в 1960-ые, являются циклизация D-глюкозы в основной или кислой среде, представленная ниже
Циклизация в основной среде
4: 6-0-тозил-D-глюкопираноза
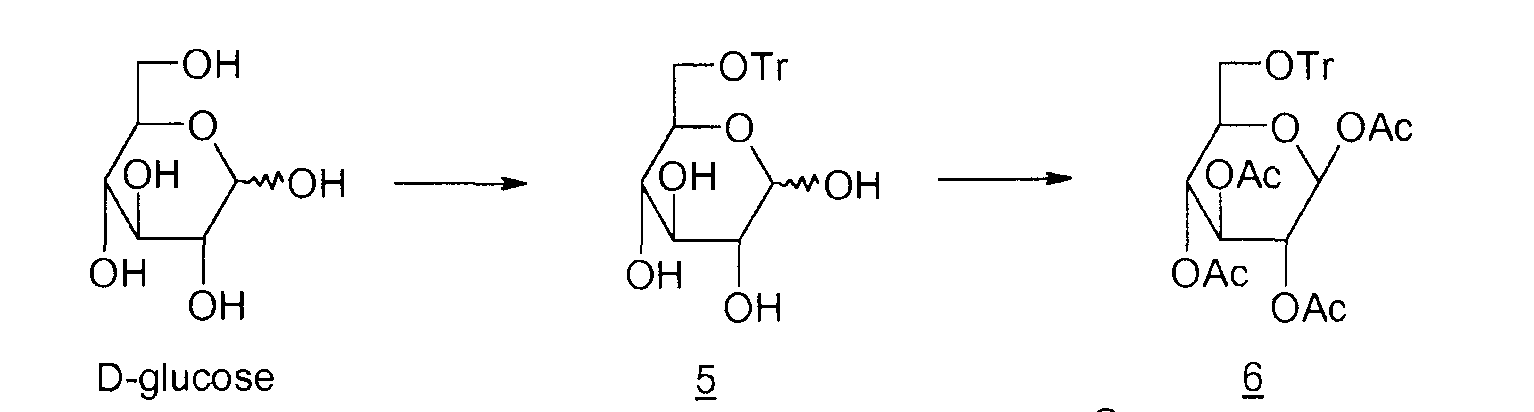
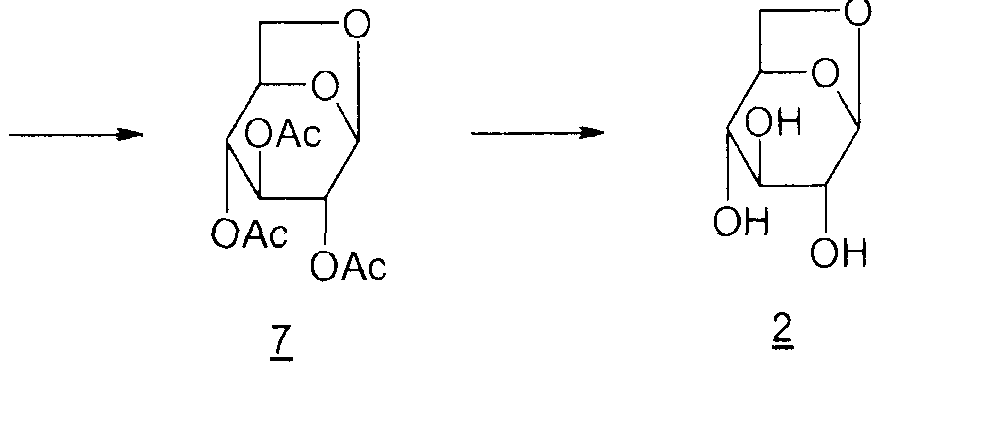
Циклизация в кислой среде
5: 6-О-тритил-D-глюкопираноза
6: 1,2,3,4-тетра-O-ацетил-6-O-тритил-β-D-глюкопираноза
7: 1,6-ангидро-2,3,4-три-O-ацетил-β-D-глюкопираноза
Циклизация в основной среде (M.A. Zottola et al., J. Org. Chem., 1989, vol. 54, p.6123-6125 ; M. Akagi et al., Chem. Pharm. Bull., 1962, vol. 10, p.905-909) выражается в низком выходе (15%). Кроме того, необходимо ацетилировать неочищенный левоглюкозан 2 для его выделения. Что касается способа циклизации в кислой среде (M.V. Rao et al., Carbohydrate Research, 1987, vol. 162, 141-144; R.L. Wistler et al., Methods Carbohydr. Chem., 1972, vol. 6, p.411-412; E. Zara-Kaczian et al., 1982, vol. 111, no. 3, p.271-283; E. Zara-Kaczian et al., Acta Chemica Acad. Scient. Hung., 1978, vol. 96, no. 3, p.311-313), описано, что он имеет лучший выход (70%), но содержит на две стадии больше.
Три способа получения эпоксида Черни (I) из соединения 1 следующие.
Способ 1:
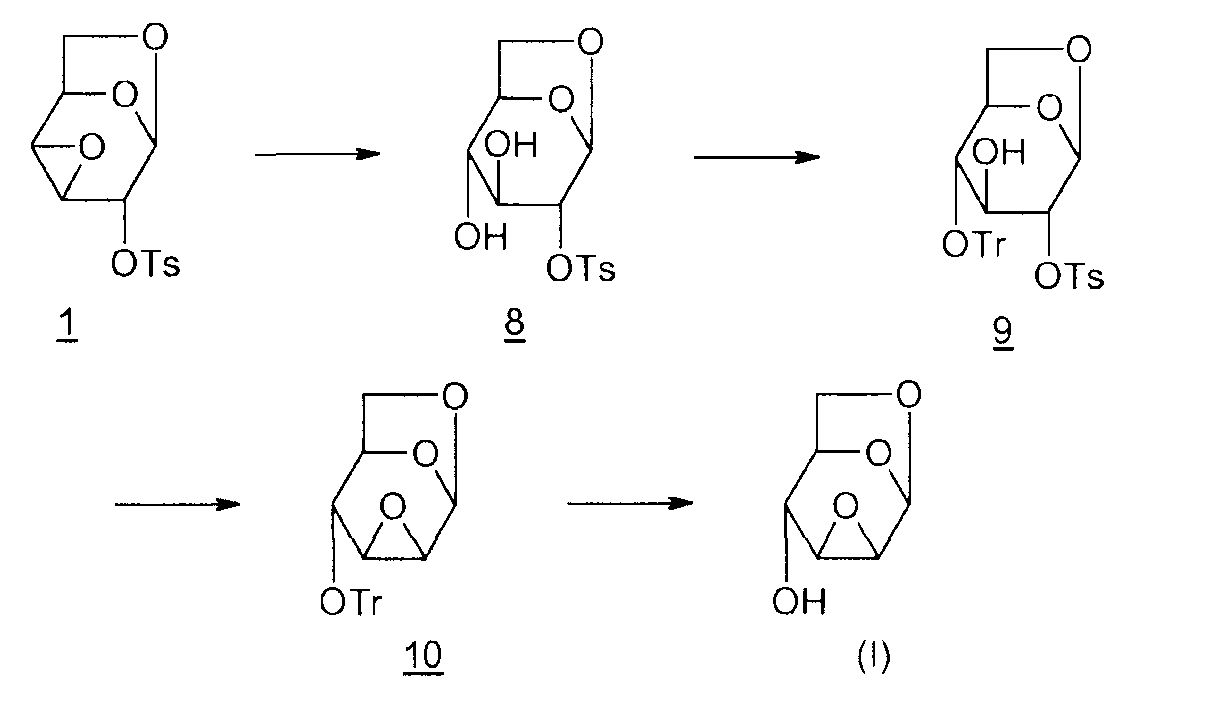
8: 1,6-ангидро-2-O-тозил-β-D-глюкопираноза
9: 1,6-ангидро-2-O-тозил-4-O-тритил-β-D-глюкопираноза
10: 1,6:2,3-диангидро-4-O-тритил-β-D-глюкопираноза
Наряду с последовательностью этапов, необходимых, чтобы получить эпоксид Черни (I), в этой цепочке трудно, кроме прочего, провести селективный гидролиз группы ангидро-3,4 на первом этапе. Гидроксил в положении 4 монотозильного производного 8 защищают затем тритильной группой (Tr), чтобы предотвратить миграцию эпоксида во время циклизации в присутствии этилата натрия (EtONa).
Способ 2:
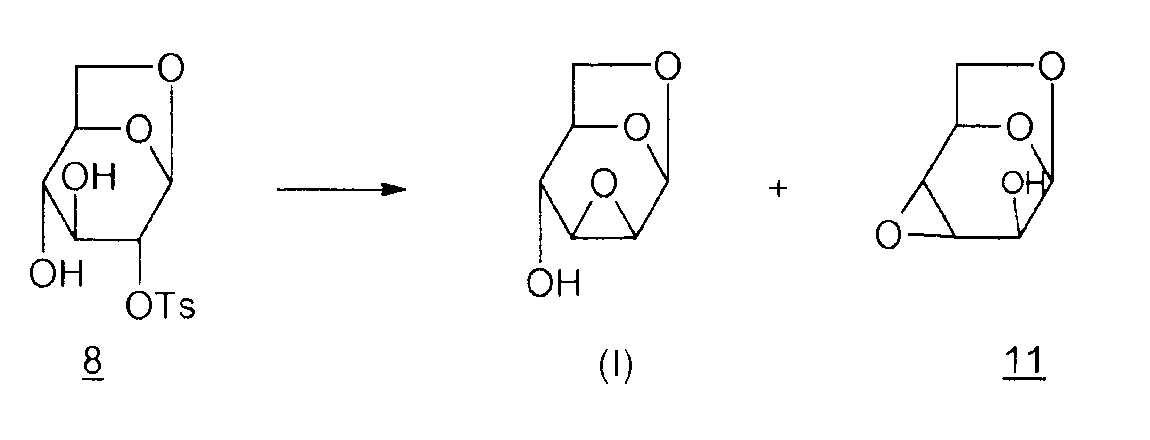
Согласно M. Cerny et al. (Synthesis, 1972, 698-699), эпоксид Черни (I) может быть получен из производного 8 в присутствии смолы амберлит IRA 400/OH-. Однако продолжительный контакт со смолой приводит к миграции эпоксида в положение 3,4 и образование производного 11 (1,6:3,4-диангидро-β-D-альтропираноза). Таким образом, остается сложным получить избирательно соединение (I). Также трудно избирательно получить исходное соединение 8, как упоминалось ранее.
Способ 3
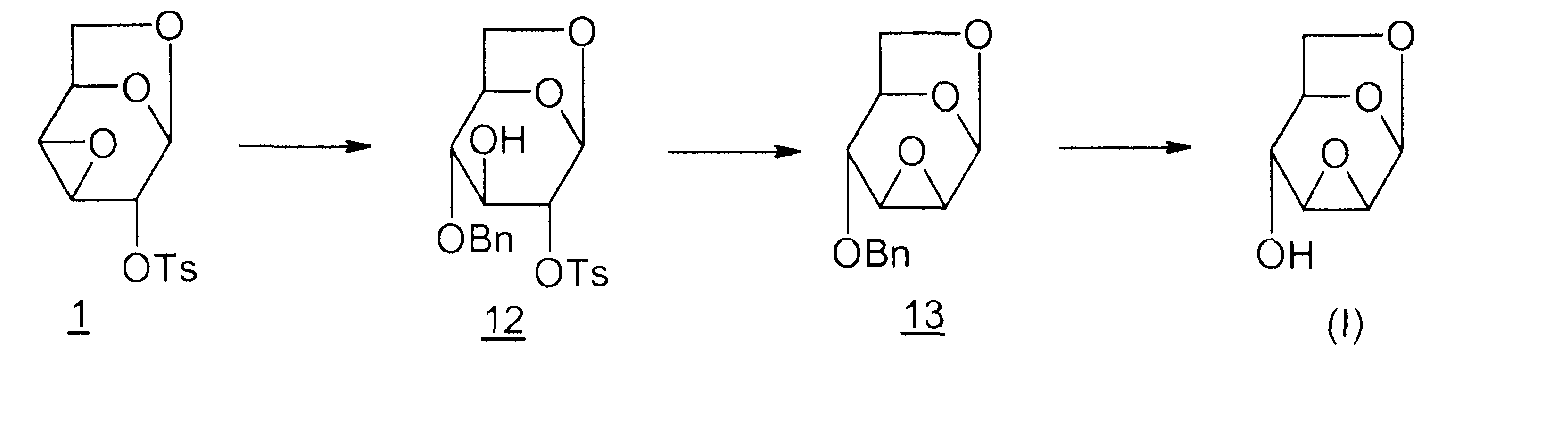
12: 1,6-ангидро-4-O-бензил-2-O-тозил-β-D-глюкопираноза
13: 1,6:2,3-диангидро-4-O-бензил-β-D-маннопираноза
Этот вариант позволяет провести циклизацию в диангидро производное 13 без миграции эпоксида (T. Trnka et al., Collect. Czech. Chem. Commun., 1971, vol. 36, p.2216-2225; M. Cerny et al., Collect. Czech. Chem. Commun., 1968, vol. 33, p.1143-1156). Тем не менее, он содержит большое число этапов получения эпоксида Черни (I) из D-глюкозы.
В заключение отметим, что три описанных выше маршрута получения эпоксида Черни (I) насчитывают соответственно 10, 8 и 9 этапов, если исходить из D-глюкозы (используя для получения левоглюкозана 2 циклизацию в кислой среде, что, как описано, является способом с наилучшим выходом), и полный выход для способов 1, 2 и 3 равен 0,5%, 10% и 13%, соответственно.
Кроме того, V. Bailliez et al. описали в Synthesis, 2003, No. 7, 1015-1017 способ получения 1,6:3,4-диангидро-β-D-альтропиранозы, который сопровождается поточным образованием 1,6:2,3-диангидро-β-D-маннопиранозы в качестве примесного продукта. Согласно этим авторам, эпоксид Черни может быть получен из предшественника, предварительно циклизованного между положениями 1 и 6, или же (N-1)-ый предшественник эпоксида Черни, ацетилированный в положении 4, может быть получен, с уровнем выхода 5%, за несколько этапов, исходя из 1,3,4-три-O-ацетил-2,6-ди-O-тозилглюкозы, подвергнутой действию оксида алюминия, микроволновому облучению и пер-O-ацетилированию.
Учитывая трудозатраты и расходы на исходные материалы, и для получения соединения (I) в промышленном масштабе необходимо разработать более короткий и, следовательно, более рентабельный синтез. Так, авторы настоящего изобретения нашли способ получения соединения (I) в два этапа, исходя из D-глюкозы, который удовлетворяет указанным выше потребностям.
Способ согласно изобретению включает в себя этапы, представленные ниже на схеме 1.
Схема 1:
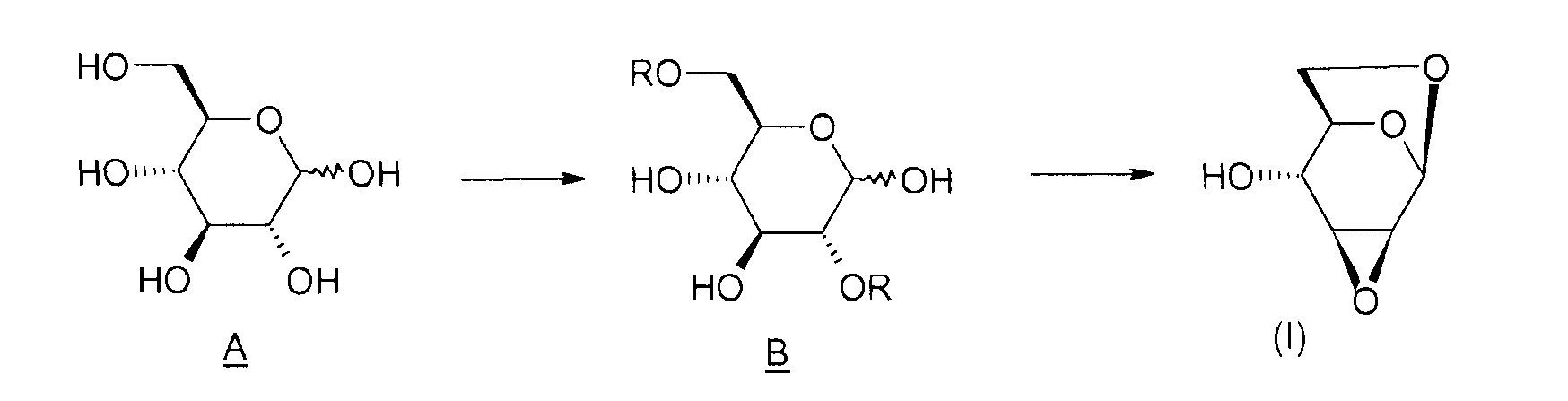
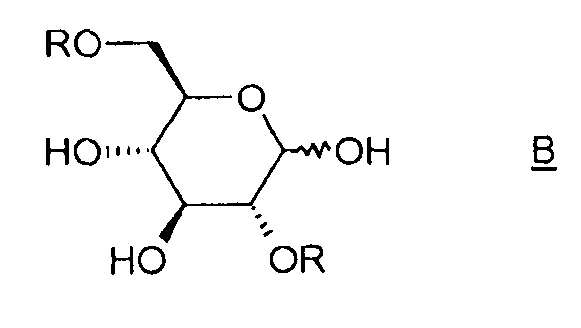
Таким образом, объектом изобретения является способ получения соединения (I), отличающийся тем, что он содержит этап циклизации соединения B, в котором R означает активатор, в присутствии основания.
Под "активатором" понимается агент, позволяющий убрать уходящую группу -OR и облегчающий реакцию циклизации между положениями 1 и 6 соединения B, например, тозил-, бензил-, бензолсульфонилгалогенид или галогенид бензолсульфонильного производного, такой как п-галогенобензилсульфонилгалогенид. Таким образом, соединение B таково, что R означает тозильную, бензильную, бензолсульфонильную или п-галогенобензилсульфонильную группу.
В случае тозилхлорида (TsCl) группа -OTs является отличной уходящей группой. Таким образом, выгодно использовать тозилхлорид в качестве активатора в растворителе, таком как пиридин.
Основание, используемое на определенном выше этапе циклизации, выбрано из гидроксидов аммония и неорганических оснований. Подходящими неорганическими основаниями могут быть сильные неорганические основания (например, гидроксид натрия или калия), слабые неорганические основания, в частности, твердого типа (например, карбонат калия, натрия или цезия).
Под "гидроксидом аммония" понимается соединение формулы
N+(R1)(R2)(R3)(R4)OH-, в которой R1, R2, R3 и R4, одинаковые или разные, означают алкильные группы, причем указанные алкильные группы являются линейными или разветвленными насыщенными алифатическими группами, содержащими от 1 до 4 атомов углерода. Гидроксид аммония, используемый в реакции циклизации соединения B, может состоять, например, из гидроксида тетрабутиламмония.
Этап циклизации соединения B проводится в подходящем растворителе, выбираемом в зависимости от природы используемого основания, в зависимости от познаний специалиста в этой области. Реакция может быть проведена, например, в изопропаноле, дихлорметане или ацетонитриле, или же в бинарной смеси этих растворителей.
В качестве растворителя используют, например, смесь изопропанол/дихлорметан, содержащую, например, около 5 объемных % дихлорметана, когда применяющимся основанием является гидроксид тетрабутиламмония. В этом случае реакция циклизации благоприятно реализуется при низкой температуре, в частности, при температуре меньше или равной 0°C, причем присутствие дихлорметана позволяет растворить соединение B при низкой температуре. Можно, например, осуществить реакцию циклизации при температуре в интервале от -10°C до 0°C, например, при примерно -5°C.
Когда основанием, используемым в реакции циклизации, является карбонат цезия, предпочтительно в качестве растворителя использовать ацетонитрил при температуре около 40°C.
Как известно специалисту, температура реакционной среды на этапе циклизации подбирается в зависимости от используемой пары растворитель/основание, чтобы оптимизировать кинетику реакции.
Согласно изобретению, соединение (I) получается с селективностью 65-85%, если исходить из интермедиата B. Химический выход на этом этапе, рассчитанный по выделенному продукту, составляет по меньшей мере примерно 60%.
Объектом изобретения является также способ получения соединения (I), отличающийся тем, что он содержит этап активации соединения A (D-глюкоза), позволяющий получить соединение B, затем этап циклизации соединения B в присутствии основания, какое определено ранее.
Этап активации соединения A может быть осуществлен с помощью активатора, какой определен ранее. Этот этап благоприятно проводить с помощью тозил-, бензил-, бензолсульфонилгалогенида или галогенида бензолсульфонильного производного, такого как п-галогенобензилсульфонилгалогенид, в растворителе, таком как пиридин.
Изобретение иллюстрируется в следующих примерах, которые детализируют способ получения соединения (I) согласно изобретению, следуя приведенной ниже схеме 2.
Схема 2:
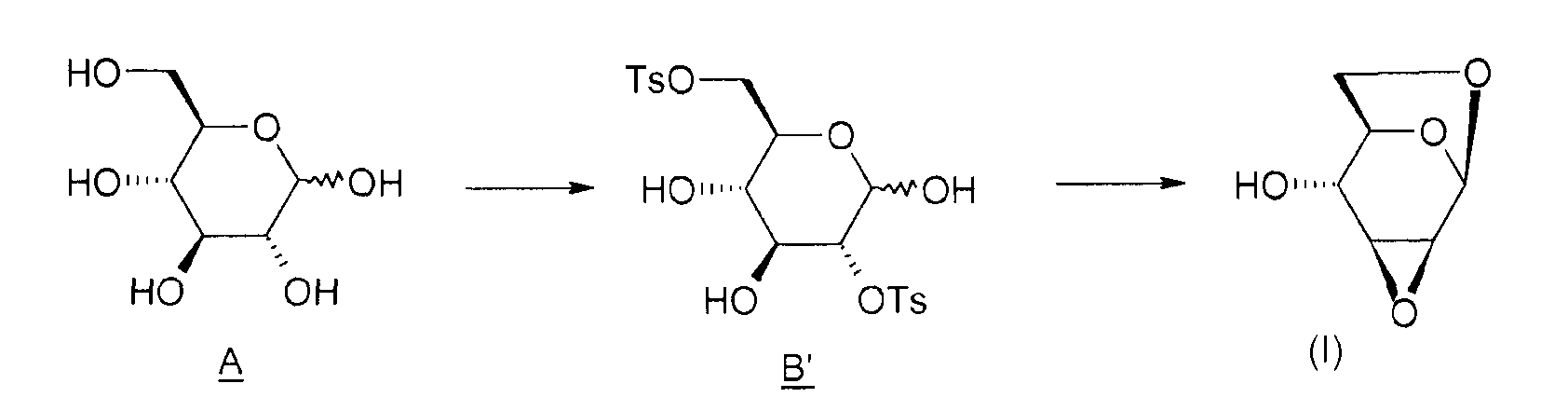
1) Получение соединения B' (2,6-ди-O-тозил-глюкопираноза)
В реактор на 2 литра, оборудованный системой перемешивания, помещают 100 г (0,55 моль) D-глюкозы и 500 г пиридина. Реакционную среду охлаждают до -10°C. В другом реакторе на 1 литр готовят раствор тозилхлорида: вводят 212 г (1,12 моль) тозилхлорида и 667 г пиридина и перемешивают при 20°C до полного растворения. Постепенно (за 4-5 ч) переносят содержимое однолитрового реактора в двухлитровый реактор, поддерживая температуру -10°C. Промывают 39 граммами пиридина и продолжают перемешивать реакционную среду в течение 17 ч при -11°C.
Проводят замену растворителя путем перегонки с помощью деминерализованной воды. Пиридина в реакционной среде должно быть ≤20%; если это не так, снова проводят перегонку с 400 мл воды.
Реакционную среду охлаждают до 20°C и вводят 400 мл деминерализованной воды. После удаления монотозильного соединения вводят 400 мл дихлорметана, 48 г соляной кислоты и 33 мл воды. Выдерживают 30 мин и измеряют pH, который должен быть меньше или равен 1; если это не так, по каплям добавляют соляную кислоту до получения pH≤1. Затем промывают раствором хлорида натрия (400 мл воды + 40 г хлорида натрия) до получения pH примерно 5-5,5.
Наконец, на роторном испарителе концентрируют фазу дихлорметана. Получают концентрат со 150 мл дихлорметана, который снова концентрируют. После проведения трех таких операций получают соединение B' в форме бежевой (кремовой) пены.
Ожидаемая масса продукта=271 г
Полученная масса продукта=184 г
Органическая чистота=81,6%, измеренная по ВЭЖХ (высокоэффективная жидкостная хроматография)
Химический выход=55%
2) Получение соединения (I)
2.1: Циклизация, осуществленная с помощью гидроксида тетрабутиламмония
В реактор объемом 1 литр вводят 10 г соединения B', полученного на предыдущем этапе, 100 мл изопропанола и 5 мл дихлорметана. Реакционную среду охлаждают до температуры -5°C при перемешивании со скоростью 400 об/мин. Медленно вливают (в течение примерно 30 мин) 26,6 г гидроксида тетрабутиламмония (40% в воде). Оставляют перемешиваться на 30 минут и реакцию останавливают, нейтрализуя реакционную среду 12%-ной соляной кислоты, до получения pH примерно 6-7. Меняют растворитель (замена изопропанола на этилацетат) на роторном испарителе с 6 мл этилацетата. Расчетный химический выход на этом этапе составляет около 60%, органическая чистота соединения (I), измеренная, исходя из пиков газовой хроматографии, равна 68%.
Спектры ЯМР H' и С-13 соединения (I) сняты на приборе Bruker 300 МГц. Химические сдвиги выражены относительно тетраметилсилана, с точностью 0,01 ч/млн для протонного спектра и точностью 0,1 ч/млн для спектра С-13. Константы взаимодействия указаны в абсолютных величинах в Гц, точность 0,5 Гц.
1H-ЯМР (CDCl3): 2,67 (д, 1H, OH, J4,OH 5,5 Гц), 3,12 (д, 1H, H3, J2,3 3,4 Гц), 3,42 (дд, 1H, H2, J2,3=J2,1=3,0 Гц), 3,69-3,77 (м, 2H, H6, H6), 3,89 (д, 1H, H4, J4,OH 5,5 Гц), 4,40 (дм, 1H, H1, J1,2 3,0 Гц).
13C-ЯМР: 49,3: C3; 54,3: C2; 65,6: C6,6'; 67,1: C4; 97,7: C1; 74,2: C5.
2.2: Циклизация, осуществленная с помощью карбоната цезия
Как вариант, действуют, как указано в примере 2.1, но используя карбонат цезия в качестве основания для реакции циклизации соединения B'.
Используют 2 эквивалента карбоната цезия в расчете на количество соединения B', а именно 1,133 г карбоната цезия на 1 г соединения B', в 10,5 мл ацетонитрила и при температуре около 40°C. Химический выход, оцененный для этого этапа, составляет около 80%, причем органическая чистота полученного так соединения (I), измеренная из площади пиков в газовой хроматографии, равна 87%.
Идентификация молекул, модулирующих белок-белковое взаимодействие
Замещенные бензоиламиноиндан-2-карбоновые кислоты и родственные соединения
Замещенные n-фенилбипирролидинкарбоксамиды и их терапевтическое применение
Замещенные n-фенилбипирролидинкарбоксамиды и их применение в лечебных целях
Замещенные n-фенилпирролидинилметилпирролидинамиды и их терапевтическое применение в качестве модуляторов рецептора н3 гистамина
Замещенные n-фенил-бипирролидинмочевины и их терапевтическое применение
Спироциклические нитрилы в качестве ингибиторов протеазы
Применение расбуриказы для лечения или профилактики расстройств или косвенных осложнений на сердце, вызванных приступами ишемии или реперфузией
Циклические индол-3-карбоксамиды, их получение и их применение в качестве лекарственных препаратов
Способ идентификации агента на основе высокопроизводительного скрининга

