Результат интеллектуальной деятельности: ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОДУЦИРУЮЩАЯ АНТИОКСИДАНТНЫЙ, АНТИМИКРОБНЫЙ, АНТИТОКСИЧЕСКИЙ БЕЛОК - ЛАКТОФЕРРИН ЧЕЛОВЕКА, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И СПОСОБ ТЕРАПИИ
Вид РИД
Изобретение
Изобретение относится к области медицины, в частности токсикологии и радиологии, к лекарственным средствам на основе антиоксидантных белков и способам их применения.
Известна фармацевтическая композиция, содержащая лактоферрин человека, вызывающая в организме различные физиологические изменения, включая иммуномодулирующие эффекты, уменьшение воспалительных реакций и ингибирование роста солидных опухолей. (Патент США №6333311).
Известна фармацевтическая композиция, содержащая рекомбинантный лактоферрин человека (Патент США №5955316). Известна композиция, предназначенная для лечения диабетических язв при аппликационном, пероральном или парентеральном применении (Патент США №7524814).
Известен способ профилактики и лечения тяжелых послеоперационных осложнений (гнойно-воспалительных и постгеморрагических) с явлениями полиоргаиной недостаточности и общей интоксикации путем ежедневного внутривенного, внутриполостного, интратрахеального введения лекарственных препаратов, содержащих лактоферрин человека и церулоплазмин человека (Патент РФ №2199337).
К причинам, затрудняющим достижение стойкого терапевтического эффекта при использовании всех указанных композиций относится наличие в них в качестве активных компонентов выделенных и очищенных белков, которые при любых способах введения быстро выводятся из организма больного, что обусловливает необходимость многократного введения. фармацевтических композиций, содержащих антиоксидантный белок лактоферрин, для поддержания терапевтически эффективной концентрации в организме. Помимо сложности с медицинской точки зрения, это экономически невыгодно.
Таким образом, в данной области возникла острая необходимость в разработке фармацевтических композиций и способов профилактики и лечения на их основе, которые были бы терапевтически высоко эффективны, экономически оправданы, а также требовали меньших временных и трудовых затрат медицинского персонала при применении.
Известно активно развивающееся направление по разработке генно-терапевтических препаратов на основе рекомбинантных аденовирусов. Среди человеческих аденовирусов наиболее охарактеризованным является аденовирус человека пятого серотипа (Ад5) (Zaia J.A. The status of gene vectors for the treatment of diabetes, Cell Biochem, Biophys, 2007, v. 48(2-3), c.183-90 - Статус генетических векторов для лечения сахарного диабета).
Одними из преимуществ рекомбинантных аденовирусов является обеспечение высокого уровня экспрессии целевого гена в течение длительного времени, до 28 дней, у некоторых лабораторных животных дольше (до 60-90 дней) (S.L. Brody, R.G. Crystal, Adenovirus-Mediated in Vivo Gene Transfer, Annals new york acadmy of sciences, 1994, 716, c.90-103 - Обеспечиваемый аденовирусами перенос генов in Vivo).
Известно успешное применение вещества на основе рекомбинантного аденовируса, экспрессирующего лактоферрин человека в опыте на мышах с раком молочной железы. После введения вещества в ткани опухоли наблюдалось замедление ее роста вследствие апоптоза клеток (Inhibition of tumor growth by recombinant adenovirus containing human lactoferrin through inducing tumor cell apoptosis in mice bearing EMT6 breast cancer, Wang J., Li Q., Ou Y., Han Z., Li K., Wang P., Zhou S., Arch Pharm Res, 2011, Vol 34, No 6, c.987-995 - Ингибирование роста опухоли с помощью аденовируса, содержащего ген человеческого лактоферрина путем индукции апоптоза опухолевых клеток у мышей с раком груди ЕМТ6).
Однако данный препарат не обеспечивает антиоксидантного, антимикробного действия лактоферрина, т.к. при описанном способе терапии обеспечивается только его противоопухолевое действие.
Известно, что аденовирус может эффективно экспрессировать лактоферрин человека вокруг опухолевых клеток, показывая стабильное торможение роста раковых опухолей шейки матки, усиление иммунитета организма и не имеет никаких токсических побочных эффектов (Заявка на выдачу патента №200910021705, 19.08.2009, Li Q., Li J., Han Z., Wu G., Hu J., Recombinant adenovirus carrying human lactoferrin, preparation and uses thereof, Китай).
Данный препарат также обладает узким противоопухолевым действием, так как указанный способ его применения вокруг опухолевых клеток не обеспечивает широкого антиоксидантного антимикробного и антитоксического действия лактоферрина.
Наиболее близкой фармацевтической композицией того же назначения к заявляемому изобретению по совокупности признаков является антибактериальный, антиоксидантный, детоксицирующий, иммуномодулирующий и антиканцерогенный препарат, который в качестве активного вещества содержит лактоферрин человека, выделенный из женского молока (Патент РФ №2165769, принят за прототип. Известный препарат выполнен в виде лиофилизата, содержащего от 10 до 90% лактоферрина человека. Препарат применяют для профилактики и/или лечения инфекционных, воспалительных заболеваний и токсических состояний различной этиологии.
К причинам, затрудняющим достижение стойкого терапевтического эффекта при использовании указанного препарата является необходимость многократных инфузий препарата из-за быстрого выведения лактоферрина из организма больного и, соответственно, большие затраты препарата, медицинского инструментария и времени медицинского персонала для достижения требуемого результата лечения.
Уникальность и дефицитность сырья - женского молока серьезно ограничивает масштабирование производства препарата и, соответственно, возможность его применения в требуемых количествах при медицинских показаниях.
Задачей данного изобретения является создание фармацевтической композиции, продуцирующей антиоксидантный, антимикробный, антитоксический белок - лактоферрин человека, обеспечивающей достижение стойкого терапевтического эффекта при однократных введениях этой композиции, сокращение затрат препарата, медицинского инструментария и времени медицинского персонала для достижения требуемого результата лечения, способа ее получения и способа терапии посредством воздействия этой фармацевтической композицией на организм человека.
Поставленная задача решается за счет того, что, фармацевтическая композиция, в которой лечебный эффект получают в результате воздействия антиоксидантного, антимикробного, антитоксического белка лактоферрина человека, на организм человека содержит нереплицирующиеся наночастицы на основе генома аденовируса человека 5-го серотипа со вставкой гена лактоферрина человека, экспрессирующие лактоферрин человека в терапевтически эффективном количестве в организме, при этом она содержит содержащая формулирующий буфер. Содержание нереплицирующихся наночастиц составляет не менее 2,33×1011 физических частиц на мл формулирующего буфера. Способ получения фармацевтической композиции, содержащей нереплицирующиеся наночастицы на основе генома аденовируса человека 5 серотипа со вставкой экспрессирующей кассеты, включающей промотор, ген лактоферрина человека и сигнал полиаденилирования, заключается в том, что заданную активность получают путем засева пермиссивной клеточной культуры 293 нереплицирующимися наночастицами с геном лактоферрина человека и наращивания нереплицирующихся наночастиц в клетках до нужного содержания, после проводят многостадийную очистку путем центрифугирования, четырехкратного перемораживания-оттаивания в буферном растворе полученной на предыдущей стадии твердой части, дополнительно обрабатывают нуклеазой с дальнейшим отделением нереплицирующейся наночастицы с геном лактоферрина человека от разрушенных клеток центрифугированием и последующим отбором полученного супернатанта, при этом дальнейшую очистку проводят ультрафильтрацией, для чего полученный супернатант разводят буфером и перемешивают, затем полученный раствор фильтруют под давлением, и проводят очистку путем анионно-обменной хроматографии и далее экслюзионной хроматографии, к полученному элюату добавляют этанол и этилендиаминтетрауксусную кислоту, после чего отправляют на нормальную фильтрацию, затем получают готовый препарат путем разбавления полученного на предыдущей стадии препарата формулирующим буфером до заданного содержания нереплицирующихся наночастиц. При этом конструирование нереплицирующихся наночастиц со вставкой гена лактоферрина человека осуществляют методом гомологичной рекомбинации в культуре клеток. Терапевтически эффективную дозу нереплицирующихся наночастиц берут на 3 мл конечного объема композиции. Форма выпуска препарата - 1 мл, 2 мл. и 3 мл. Способ терапии, заключается во введении человеку заявленной фармацевтической композиции, продуцирующей антиоксидантный, антимикробный, антитоксический белок - лактоферрин человека. Фармацевтическая композиция вводится человеку внутривенно. Фармацевтическая композиция может вводится человеку внутривенно капельно. Введение фармацевтической композиции проводят для лечения токсических состояний, обусловленных гнойно-воспалительными заболеваниями индуцированными различными микроорганизмами, физическими воздействиями и химическими агентами, в том числе средствами лекарственной и лучевой терапии. Терапевтически эффективная доза нереплицирующихся наночастиц со вставкой гена лактоферрина человека составляет от 7×1011 физических частиц до 7×1013 физических частиц на человека. Введение фармацевтической композиции проводят последовательно в два этапа. При этом введение фармацевтической композиции на втором этапе проводят через сутки после первого введения. Введение фармацевтической композиции на первом этапе осуществляют в объеме, содержащем 1/3 полной терапевтической дозы фармацевтической композиции, а на втором этапе вводят оставшиеся 2/3 полной терапевтической дозы композиции. 1/3 полной дозы фармацевтической композиции растворяют в 66 мл физиологически приемлемого растворителя, а 2/3 полной дозы растворяют в 134 мл физиологически приемлемого растворителя. Физиологически приемлемым растворителем является раствор глюкозы. Раствор глюкозы является 5%, либо 10%.
Реализация изобретения.
Указанные единые технические, лечебные и экономические результаты при осуществлении изобретения по заявляемому изобретению достигаются за счет того, что заявляемый способ, так же как известный способ профилактики и/или лечения воспалительных, и/или инфекционных заболеваний, и/или токсических состояний осуществляют при помощи антиоксидантного белка лактоферрина. Особенность заявляемого способа заключается в том, что антиоксидантный белок продуцируется непосредственно в организме человека при введении нереплицирующихся наночастиц со вставкой гена антиоксидантного белка, а не вводятся многократно в виде выделенных из природного источника белков. Экспрессированный белоки оказывает лечебное действие в качестве антимикробного, и/или противовоспалительного, и/или иммуномодулирующего, и/или антиокидантного, и/или детоксицирующего, и/или антиканцерогенного агента.
Для осуществления лечебного процесса фармацевтическую композицию на основе нереплицирующихся наночастиц, продуцирующую антиоксидантный белок человека лактоферрин, вводят в следующих дозах:
- для лечения токсических состояний, обусловленных гнойно-воспалительными заболеваниями индуцированными различными микроорганизмами, физическими воздействиями и химическими агентами, в том числе средствами лекарственной и лучевой терапии, от 7×1011 ф.ч. до 7×1013 ф.ч. на человека;
- для профилактики постинъекционных осложнений в начале терапии на первом этапе 1/3 полной терапевтической дозы нереплицирующихся наночастиц с геном лактоферрина человека;
- для профилактики постинъекционных осложнений в начале терапии на втором этапе вводят 2/3 полной терапевтической дозы нереплицирующихся наночастиц с геном лактоферрина человека;
Сущность изобретения заключается в следующем. Заявляемая фармацевтическая композиция содержит в качестве основного действующего компонента нереплицирующиеся наночастицы со вставкой гена, экспрессирующего антиоксидантный белок человека - лактоферрин. Этот белок проявляет в организме человека многогранные биологический свойства, которые обусловливают его антиоксидантное, детоксицирующее, иммуномодулирующее, антимикробное, противовоспалительное и антиканцерогенное действие. Основным существенным отличием заявляемой фармацевтической композиции от ранее известных фармацевтических композиций является то, что антиоксидантный белок длительное время продуцируется при однократном введении наноструктуры непосредственно в организме, а не вводится многократно в организм в виде выделенного из какого-либо источника белка. Фармацевтическая композиция совместима с любыми фармацевтически пригодными растворителями. Заявляемые пределы количества фармацевтической композиции, вводимой в организм больного, позволяют сохранять высокую терапевтически эффективную концентрацию целевого антиоксидантного белка в организме в течение длительного времени.
Фармацевтическая композиция является исходным продуктом для приготовления различных лекарственных форм, применение которых определяется в зависимости от заболевания. Заявляемая фармацевтическая композиция на основе нереплицирующихся наночастиц со вставкой гена, кодирующего антиоксидантные белки человека, прошла доклинические и клинические испытания по изучению специфической (терапевтической) эффективности и общетоксического действия, которые показали безвредность данной композиции и терапевтическую активность в качестве антиоксидантного, антибактериального и детоксицирующего вещества, что иллюстрируется приведенными ниже примерами.
Для создания фармацевтичекой композиции, содержащей в своем составе нереплицирующиеся наночастицы на основе генома аденовируса человека 5-го серотипа и пролонгированно экспрессирующей лактоферрин человека в организме человека в терапевтических количествах необходимо:
1) сконструировать нереплицирующиеся наночастицы со вставкой гена лактоферрина;
2) разработать способ получения фармацевтической композиции;
3) доказать соответствие экспрессирующегося нереплицирующимися наночастицами лактоферрина человека нативному лактоферрину человека;
4) определить путь введения, терапевтическую дозу и допустимые пределы доз;
5) доказать пролонгированность действия фармацевтической композиции;
6) разработать способ безопасного и терапевтически эффективного применения фармацевтической композиции.
Приведеные далее примеры, таблицы и рисунки раскрывают сущность изобретения и подтверждают эффективность решения по данному изобретению.
Пример 1.
1) Конструирование нереплицирующейся наночастицы на основе генома аденовируса человека 5 серотипа со вставкой гена лактоферрина человека.
2) Получение фармацевтической композиции.
Конструирование нереплицирующейся наночастицы на основе генома аденовируса человека 5 серотипа (размером 70-80 нм) со вставкой гена лактоферрина человека осуществляли методом гомологичной рекомбинации в клеточной культуре и проводили с использованием общеизвестных лабораторных методик (например, Сэмбрук Дж., Фрич Э., Маниатис Т. и др. Методы генетической инженерии. Молекулярное клонирование. М., Мир, 1984, с.205-224, 387-420). За основу была взята рекомбинантная плазмида pJM17 (W.J. McGrory, D.S. Bautista, F.L. Graham. A simple technique for the rescue of early regionl mutations into infectious human adenovirus type 5, Virology, V, 163, №2, 1988, с.614 - Простая техника для удаления раннего региона 1 в инфекционном аденовирусе человека 5 типа), с делециями в области Е1 аденовирусного генома. Искусственно синтезированный кДНК гена лактоферрина человека лигировали в общеизвестную шаттл-плазмиду pRcCMV (Invitrogen, San Diego, CA, №V75020). Далее для взаимной трансформации полученной плазмиды pRcCMV - Lf и плазмиды pJM17 ими трансфецировали клетки 293 (например, №300192, CLS, Germany) с помощью метода кальциево-фосфатной преципитации (Graham FL, van der Eb AJ, 1973-A new technique for the assay of infectivity of human adenovirus 5 DNA". Virology, 52 (2) 456-67 - Новая техника метода заражения ДНК аденовируса человека 5 серотипа). В результате получили нереплицирующиеся наночастицы, содержащие экспрессирующую кассету с CMV-промотором, геном лактоферрина человека и сигналом полиаденилирования. Бляшки рекомбинантной частицы образовывались на культуре клеток через несколько дней после трансфекции, их отбирали пастеровской пипеткой, полученный материал размножали на клетках линии 293 до получения титра 3×1010 ф.ч. (физических частиц)/мл (108 ЕД/мл).
Получение фармацевтической композиции.
Необходимое содержание нереплицирующихся наночастиц в композиции было определено в примерах 4 и 5 по антитоксическому эффекту и должно составлять не менее 2,33×1011 ф.ч./мл (что соответствует активности не менее 6,7×108 ЕД/мл). Объем препарата должен составлять 3 мл, что связано с удобством фасовки и применения. Получение данной композиции проходит в несколько этапов.
Полученную выше клеточную суспензию, содержащую нереплицирующиеся наночастицы в титре 3×1010 ф.ч./мл, использовали для дальнейшего наращивания титров нереплицирующихся наночастиц и приготовления готовой фармацевтической композиции с заданным содержанием не менее 2,33×1011 ф.ч./мл (что соответствует активности не менее 6,7×108 ЕД/мл).
Для наработки необходимых титров нереплицирующихся наночастиц волновой биореактор с 4500 мл суспензии пермиссивной клеточной культуры 293 засевали клеточной суспензией объемом 500 мл, содержащей нереплицирующиеся наночастицы с титром 3×1010 ф.ч./мл.
Культивировали для наращивания нереплицирующихся наночастиц внутри клеток и достижения их содержания 6×1010 ф.ч./мл (активность 2×108 ЕД/мл), ориентировочно в течение 48 часов. По достижению необходимого содержания наночастиц клеточную массу подавали на очистку, которая состояла из нескольких стадий:
1) Проводили осаждение клеточной массы центрифугированием. Поступающая на очистку суспензия имела не менее 1014 ф.ч. на 5 л (оценивали при помощи масс-спектрометра, 1 ОЕ=1012 ф.ч.). Центрифугирование проводили при режиме 6000 g в течение 15 мин, при этом жидкий надосадок сливали, а оставшуюся твердую часть, содержащую клетки и нереплицирующиеся наночастицы подавали на дальнейшие стадии очистки.
2) Извлечение нереплицирующихся наночастиц из клеточной культуры проводили путем разрушения клеток четырехкратным перемораживанием-оттаиванием. Готовили буферный раствор с pH 8.0: 5 mM ТрисHCl, 0.075 MNaCl, 1 mM MgCl2) 5% сахароза, 1% полисорбат 80. Полученный в предыдущую стадию осадок ресуспендировали в 70 мл буфера (коэффициент содержания ×71).Объем раствора составлял 80 мл.
Замораживание проводили в течение 2 часов в жидком азоте, размораживали на водяной бане (при +37°C), не допуская перегрева.
3) Для облегчения дальнейшего удаления геномной клеточной ДНК проводили дополнительную обработку нуклеазой. Для этого добавляли бензоназу до концентрации в растворе 150 U/мл и ставили на мягкое перемешивание с помощью магнитной мешалки на 3 часа при комнатной температуре (21-23°C).
4) Отделение нереплицирующихся наночастиц от разрушенных клеток осуществляли центрифугированием при 9000 g 10 мин. Отбирали супернатант, содержащий нереплицирующиеся наночастицы.
5) Дальнейшую очистку проводили ультрафильтрацией. Для этого полученный супернатант разводили буфером (50 mM TrisHCl pH 7.5, 1М NaCl, 2 mM MgCl2, 5% сахароза, pH 7,5) до объема не менее 200 мл, перемешиваем с помощью магнитной мешалки. В процессе фильтрации объем циркулирующего раствора (ретентата) постоянно доводили до исходного (200 мл).
6) Далее очистку производили путем анион-обменной хроматографии.
Ретентат наносили на колонку (AxiChrom 70/300 объемом 400 мл), содержащую анионнообменный сорбент Q Sepharose virus licenced. Нереплицирующиеся наночастицы сорбируются на колонке, в то время как примеси не сорбируются, а вымываются буфером А. После удаления примесей нереплицирующиеся наночастицы десорбировали промывкой буфером Б. Условия хроматографирования: поток 193 мл/мин, буфер A (40 mM TrisHCl, 0,27 М NaCl, 2 mM MgCl2, 5% Сахароза, 0,1% Полисорбат 80, pH 7.5), проводимость ~28-30 mS/cm; буфер Б (40 mM TrisHCl, 0.5М NaCl, 2 mM MgCl2, 5% сахароза, 0.1% полисорбат 80, рН 7.5) проводимость ~50 mS/cm. Элюат в объеме 200 мл отправляли на следующую стадию.
7) Эксклюзионная хроматогафия
Поученный в предыдущей стадии элюат наносили на колонку (AxiChrom 100/300 объемом 800 мл), содержащую сорбент Q Sepharose 4 FastFlow. Высокомолекулярные вещества, не входящие в поры сорбента, элюировали первым пиком (к ним относятся нереплицирующиеся наночастицы), примеси элюировали после выхода пика нереплицирующихся наночастиц. Условия хроматографирования: поток 130 мл/мин, буфер (10 mM TrisHCl, 75 мМ NaCl, 1m MMgCl2, 5% сахароза, 0,05% полисорбат 80, pH 8.0).
К полученномуй элюату (80 мл) добавляли этанол до концентрации 0,5% и этилендиаминтетрауксусную кислоту (ЭДТА) до концентрации 100 мкМ, отправляли на следующую стадию.
8) Нормальная фильтрация.
Для стерилизации полученного препарата проводили фильтрование через систему фильтров с размером пор 22 мкМ. Конечный объем препарата на данной стадии составлял 80 мл и содержал нереплицирующиеся наночастицы в титре 1×1012 ф.ч./мл. Его разбавляли формулирующим буфером (например, 10 mM TrisHCl, 75 mM NaCl, 1 mM MgCl2, 5% сахароза, 0,05% полисорбат 80, 0,5% Этанол, 100 мкм ЭДТА, pH 8.0) до получения заданного содержания 2,33×1011 ф.ч./мл и стерилизовали нормальной фильтрацией.
Таким образом, вышеописанный способ получения фармацевтической композиции позволяет на основе сконструированной нереплицирующейся наночастицы со вставкой гена лактоферрина человека получить в препарате содержание нереплицирующихся наночастиц не менее 2,33×1011 ф.ч. на мл формулирующего буфера (что соответствует активности фармацевтической композиции не менее 6,7×108 ЕД/мл), а полная терапевтическая доза для человека (7×1011 ф.ч.) содержится в 3 мл фармацевтической композиции, что соответствует поставленной задаче.
Пример 2.
Сравнение нативного лактоферрина человека и лактоферрина человека, экспрессируемого нереплицирующимися нанаочастицами по физико-химическим свойствам и биологической активности.
Нереплицирующиеся наночастицы на основе генома аденовируса человека 5-го серотипа экспрессируют лактоферрин, называемый рекомбинантным, его соответствие нативному лактоферрину определяли по физико-химическим свойствам и биологической (антиоксидантной) активности. Для этого рекомбинантный лактоферрин человека выделяли из пермиссивной культуры клеток 293 линии, трансдуцированных нереплицирующимися наночастицами со вставкой гена, кодирующего лактоферрин человека. Очистку рекомбинантного лактоферрина человека из культуральной жидкости проводили стандартным методом аффинной хроматографии на матрице активированной CNBr сефарозы 4B, ковалентно связанной с высокоочищенными антителами к лактоферрину человека. Физико-химические свойства анализировали общеизвестными методами электорфореза, иммуноблоттинга, а биологическую (антиоксидантную) активность - известным методом ингибирования перекисного окисления липидов (ПОЛ) в гомогенате печени мышей. Нативный лактоферрин получали из донорского женского молока (Патент РФ №2165769, 13.07.2000).
1) Данные электрофоретического анализа показали, что рекомбинантный лактоферрин человека содержит белок, молекулярная масса которого составляет 76 кДа, что соответствует молекулярной массе лактоферрина человека, выделенного из донорского женского молока.
2) Методом иммуноблоттинга выявили, что рекомбинантный белок специфически взаимодействует с поликлональными антителами против лактоферрина человека (Sigma, кат. №L3262).
3) Антиоксидантная активность рекомбинантного лактоферрина человека - 1,3×10-6 моль/мл сравнима с активностью лактоферрина из женского молока - 1,4×10-6 моль/мл.
Таким образом, установлено соответствие рекомбинантного лактоферрина, экспрессированного нереплицирующимися наночастицами на основе генома аденовируса человека 5 серотипа и нативного лактоферрина из женского донорского молока по физико-химическим свойствам и антиоксидантной активности.
Пример 3.
Сравнение антимикробной активности.
Соответствие антимикробной активности рекомбинантного лактоферрина в сравнении с нативным лактоферрином, выделенным из женского молока, оценивали в отношении эталонных штаммов и клинических изолятов микрометодом в бульоне с использованием методических указаний по определению чувствительности микроорганизмов к антибактериальным препаратам (Методические указания МУК 4.2.1890-04 "Определение чувствительности микроорганизмов к антибактериальным препаратам", утв. Главным государственным санитарным врачом РФ 4 марта 2004 г.).
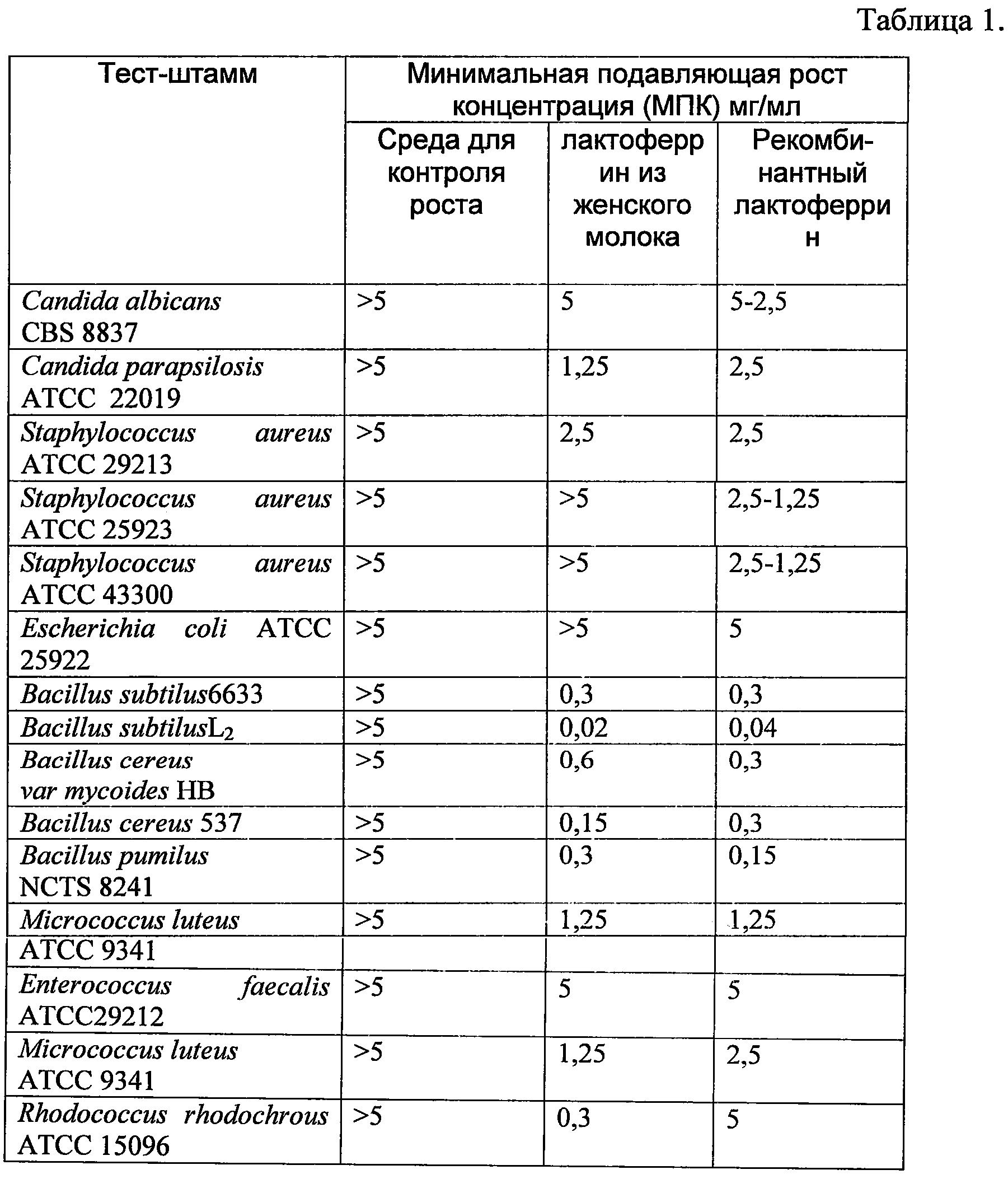
Представленные в таблице 1 данные показывают в большинстве случаев одинаковые или меньшие по рекомбинантному лактоферрину (что означает даже лучшую антимикробную активность у него по сравнению с нативным лактоферрином) значения минимальных концентраций рекомбинантного лактоферрина человека и нативного лактоферрина человека, подавляющих рост стандартных тест-микробов.
Таким образом, было установлено, что рекомбинантный лактоферрин, экспрессируемый нереплицирующимися наночастицами на основе генома аденовируса человека 5-го серотипа со вставкой гена лактоферрина человека и нативный лактоферрин, выделенный из женского молока,обладают сходной антимикробной активностью.
Пример 4.
Оценка детоксицирующего действия с определением минимальной эффективной дозы.
Детоксицирующее действие фармацевтической композиции оценивали in vivo с использованием лабораторных животных на модели токсикоза, индуцированного цитостатическим препаратом цисплатин, широко используемым в схемах полихимиотерапии больных со злокачественными процессами.
Противоопухолевое действие цисплатина реализуется за счет образования свободно радикальных продуктов, которые нарушают синтез ДНК путем внутри- и межнитевых сшивок ДНК, а также индуцируют перекисное окисление липидов (ПОЛ) клеточных мембран почек, печени, легких и других органов. Это, в конечном итоге, приводит к нарушению функций различных органов и обширному комплексу токсических реакций, интенсивность которых возрастает с увеличением дозы цисплатина.
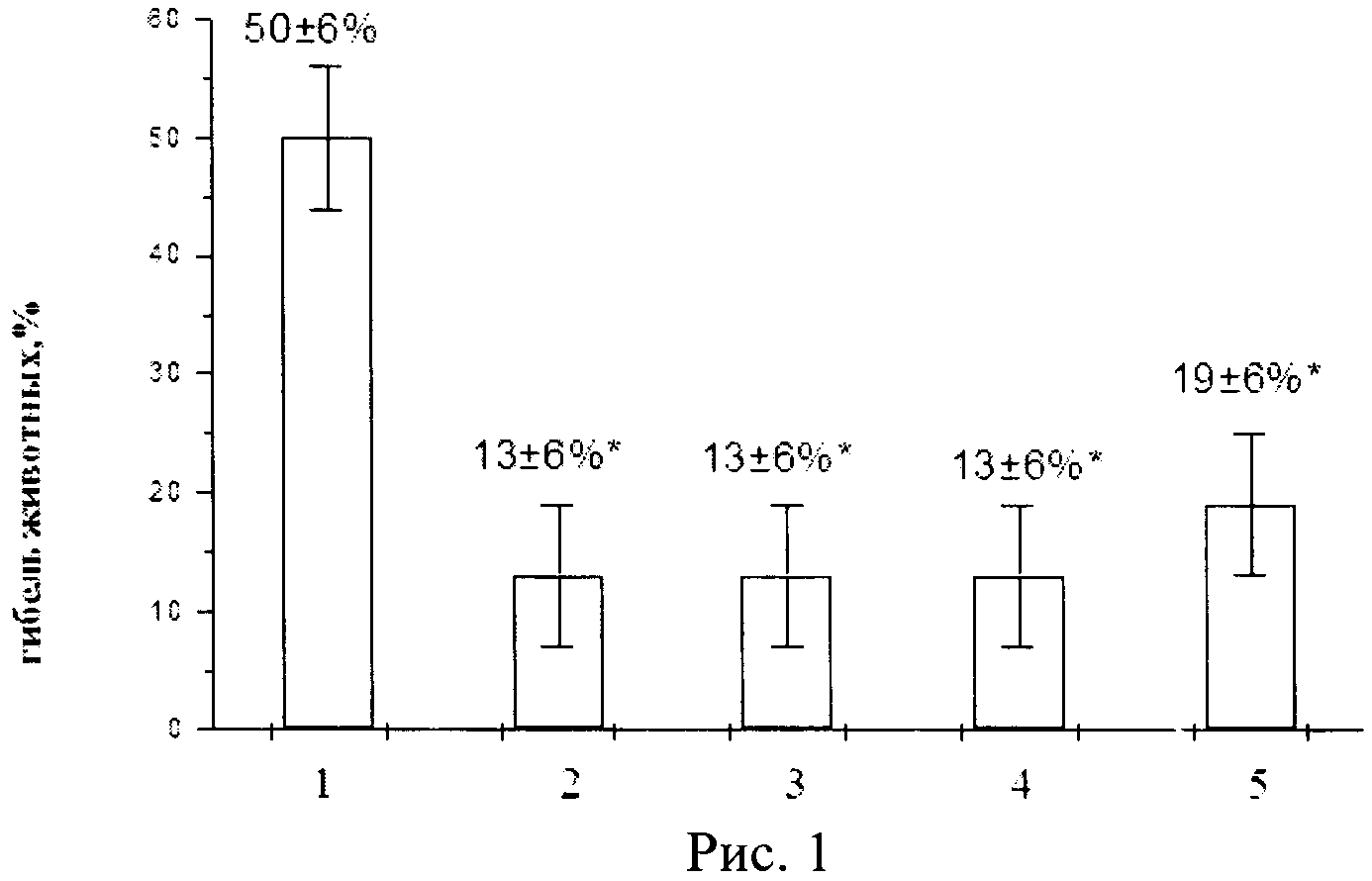
Для оценки влияния фармацевтической композиции на токсические реакции вызванные цисплатином, его однократно вводили в дозе 16 мг/кг, что приводило к 50±6%-ой гибели животных контрольной группы от острых токсических реакций (рис.1). Гибель животных начиналась с 4-х суток после воздействия и продолжалась в течение 5-ти дней. Токсические явления выражались в повышении активности ферментов - ACT и АЛТ, повышении уровня креатинина на 7-е и мочевины на 3-й и 7-е сутки наблюдения (табл.2).
В качестве детоксицирующего агента животным опытных групп за 72 часов до введения цисплатина внутривенно вводили нереплицирующиеся наночастицы со вставкой гена лактоферрина человека в дозах 1,4×1011, 4,3×1011, 4,3×1012 ф.ч./м2, а животным группы сравнения трехкратно вводилинативный лактоферрин человека из донорского женского в разовой дозе 10 мг/кг (курсовая доза 30 мг/кг), первое введение - через 24 часа после инфузии цисплатина.
На рисунке 1 представлены данные, где:
1 - гибель животных контрольной группы, цисплатин вводили однократно внутривенно в дозе 16 мг/кг;
2 - гибель животных опытной группы, которым вводили фармацевтичекую композицию однократно внутривенно в дозе 1,4×1011 ф.ч./м2 за 72 ч до введения цисплатина;
3 - гибель животных опытной группы, которым вводили фармацевтичекую композицию однократно внутривенно в дозе 4,3×1011 ф.ч./м2 за 72 ч до введения цисплатина;
4 - гибель животных опытной группы, которым вводили фармацевтичекую композицию однократно внутривенно в дозе 4,3×1012 ф.ч./м2 за 72 ч до введения цисплатина;
5 - группа сравнения, животным которой вводили нативный лактоферрин человека из донорского женского молока через 24 ч после введения цисплатина в разовой дозе 10 мг/кг в течение последующих 3-х дней (курсовая доза 30 мг/кг).
Таким образом, в период реализации токсического действия цисплатина в опытных группах животных наблюдали максимальную продукцию рекомбинантного лактоферрина человека, сохранявшуюся в течение 10-ти дней, что приводило к снижению гибели животных до 13±6% при введении нереплицирующихся наночастиц со вставкой гена лактоферрина человека в дозах 1,4×1011, 4,3×1011 и 4,3×1012 ф.ч./м2. В группе сравнения, где животные получали трехкратно нативный лактоферрин человека, летальность составила 19±6%.
Полученные результаты означают наличие детоксицирующего эффекта против токсического действия цисплатина у фармацевтической композиции, продуцирующей рекомбинантный лактферрин и нативного лактоферрина из женского донорского молока.
Результаты биохимических исследований подтвердили снижение интенсивности токсических реакций цисплатина при введении фармацевтической композиции в дозах 4,3×1011 ф.ч./м2 и 4,3×1012 ф.ч./м2.
|
Так, в таблице 2 представлены данные, которые показывают что практически все биохимические показатели крови животных в группах с введением цисплатина на фоне воздействия фармацевтической композиции и нативного белка соответствовали среднестатистическим нормальным значениям на все сроки наблюдения. Показатели функционального состояния печени - ACT и АЛТ не повышались в этих группах столь значительно, как в контрольных, а показатели, отражающие функциональное состояние почек - креатинин и мочевина - сохраняли близкие нормальным значения в отличие от достоверно повышенных значений в контрольных группах, т.е. рекомбинантный лактоферрин человека, как и нативный, выделенный из женского молока, способствует снижению нефро- и гепатотоксичности, вызываемых цитостатиком.
Таким образом, рекомбинантный лактоферрин человека обладает детоксицирующим действием в отношении токсичности цисплатина, что свидетельствует о его функциональной активности, подобной нативному белку.
Оценка детоксицирующего действия фармацевтической композиции на моделях химически индуцированного токсикоза позволила определить минимальную терапевтически эффективную дозу, равную 4,3×1011 ф.ч./м2 (токсическая доза (ТД) выражена в количестве нереплицирующихся наночастиц на м2 поверхности тела животного, дозы лекарственных средств, рассчитанные на площадь поверхности тела для различных видов животных и человека, являются эквивалентными. (Хабриев Р.У., Руководство по экспериментальному (доклиническому) изучению новых фармакологических веществ, 2000 г., 98 стр.).
Пример 5
Определение пределов переносимых доз.
Значения пределов переносимых доз фармацевтической композиции определяли в экспериментах на мышах при однократном внутривенном введении («острая» токсичность) лабораторным животным в дозах: 4,3×1011 ф.ч./м2, 43,0×1011 ф.ч./м2, 215,0×1011 ф.ч./м2, 430,0×1011 ф.ч./м2 и 860,0×1011 ф.ч./м2.
При изучении «острой» токсичности, минимальная терапевтическая доза, рассчитанная на м2, была увеличена в 10, 50, 100 и 200 раз. Превышение ТД в 10-200 раз достаточно для получения достоверной информации о токсикологической безопасности исследуемого фармакологического средства. Минимально-эффективная доза, равная 4,3×1011 ф.ч./м2 и путь введения были выбраны на основании результатов исследований, полученных в ходе изучения его фармакологической активности на животных с тяжелой экзогенной интоксикацией, вызванной введением в организм животных токсических и смертельных доз высокотоксичного противоопухолевого препарата (пример 4).
У подопытных животных после однократного внутривенного введения фармацевтической композиции в вышеуказанных дозах регистрировали клинические признаки возможной интоксикации, гибель от токсичности, сроки гибели, изменение массы тела. Взвешивание животных осуществляли до введения препарата (фон), а также на 3, 7, 10, 14, 21 и 30 сутки после введения препарата. На 30 сутки все выжившие животные были подвергнуты эвтаназии с последующей аутопсией. Проводили патологоанатомическое исследование всех трупов животных, включающее макроскопическую оценку состояния полостей организма, внутренних органов и тканей. Местнораздражающее действие препарата оценивали визуально при осмотре места инъекции. Контрольным животным вводили стабилизирующий буферный раствор (контрольное вещество №1) и изотонический (0,9%) раствор хлористого натрия (контрольное вещество №2).
Полученные данные, характеризующие токсичность фармацевтической при однократном внутривенном применении мышам, представлены в таблицах 3, 4, 5.
|
Представленные в таблице 3 результаты эксперимента показывают, что однократное внутривенное введение фармацевтической композиции мышам - самцам и мышам-самкам в дозах от 4,3×1011 ф.ч./м2 до 430×1011 ф.ч./м2 было удовлетворительно перенесено животными. Гибель от токсичности отсутствовала. При введении фармацевтической композиции в дозе, равной 860×1011 ф.ч./м2 наблюдали гибель мышей от токсического действия. Гибель в группе мышей-самцов составила 33%, а в группе мышей-самок - 17%. Средняя гибель мышей (самцов и самок) при использовании этой дозы препарата составила 25%. У погибших животных внешние проявления интоксикации отсутствовали.
|
В таблице 4 представлены о наличии интоксикации - после введения фармацевтической композиции во всех исследованных дозах у мышей (как у самцов, так и самок) наблюдали внешние проявления интоксикации в виде непродолжительного (в течение 1-го - 3-х часов) снижения двигательной активности (гиподинамия). Через 24 часа после введения и далее, в течение всего периода времени наблюдения за животными (30 суток) внешние проявления интоксикации отсутствовали. Животные были активные, реакция на человека, тактильные и болевые раздражители была выражена.
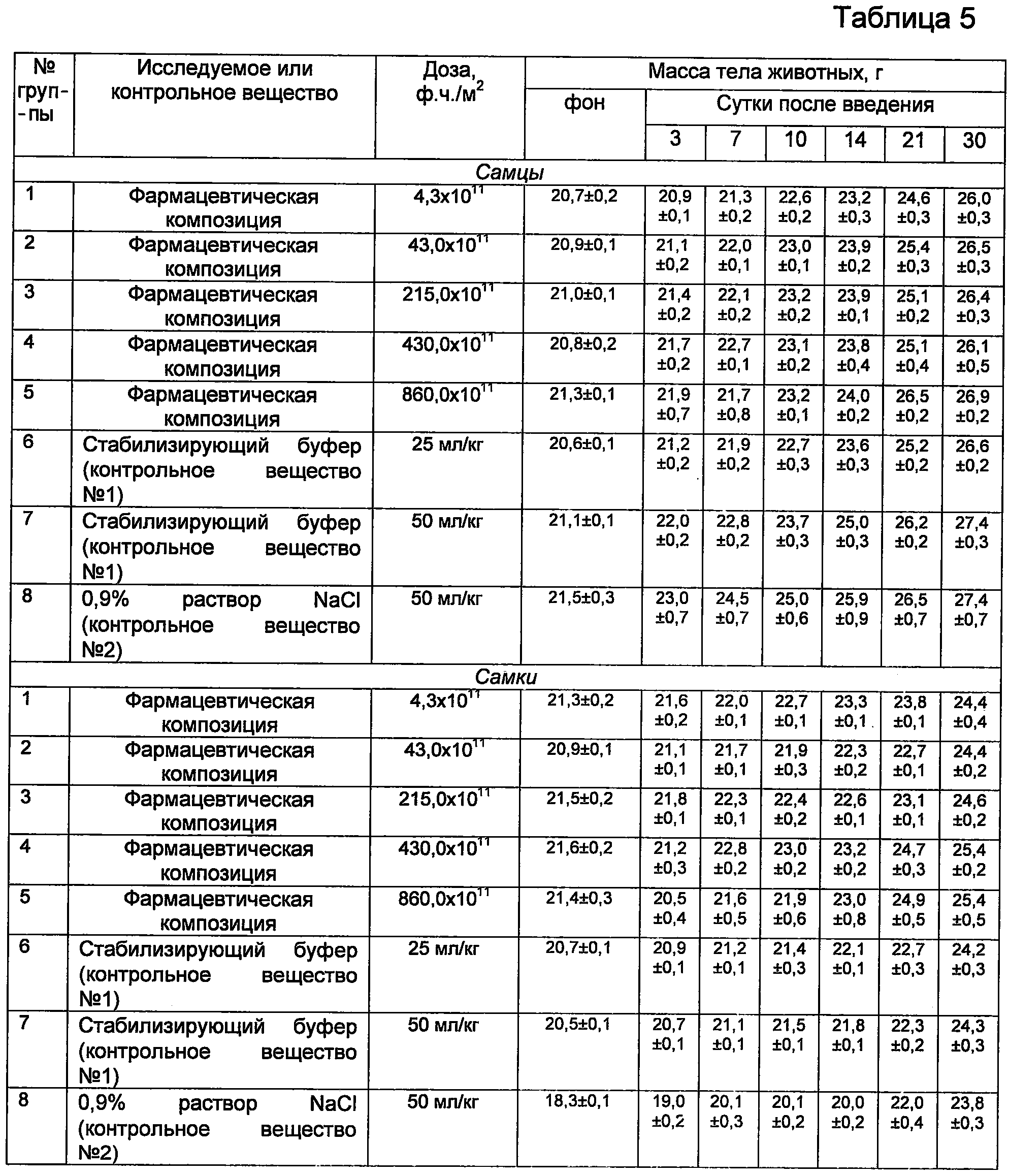
Как видно из данных, представленных в таблице 5, у мышей получавших фармацевтическую композицию однократно внутривенно в дозах от 4,3×1011 ф.ч./м2 до 860,0×1011 ф.ч./м2, а также у животных контрольных групп, получавших стабилизирующий буферный раствор и изотонический (0,9%) раствор хлористого натрия, вес (как самцов, так и самок) равномерно увеличивался на протяжении всего срока наблюдения за животными (30 суток).
Вскрытие погибших мышей выявило венозное полнокровие внутренних органов, а именно, селезенки и печени. В других органах и тканях этих мышей макроскопически патологических изменений, связанных с токсическим действием композиции не выявлено.
В результате проведенных исследований определили максимально переносимые (МПД) и летальные дозы фармацевтической композиции для мышей при его однократном внутривенном введении:
- доза 430,0×1011 ф.ч./м2 охарактеризована как МПД;
- доза 860,0×1011 ф.ч./м2 охарактеризована как частично смертельная, приводящая к гибели 25% животных.
Различий в чувствительности самцов и самок, взятых в исследование животных, к токсическому действию фармацевтической композиции при его однократном внутривенном введении не выявлено.
Внешние проявления интоксикации у мышей при использовании переносимых и максимально переносимых доз выражались в гиподинамии или адинамии, степень выраженности и длительность которых увеличивались при повышении дозы фармацевтической композиции.
Использование летальной дозы фармацевтической композиции у мышей (860,0×1011 ф.ч./м2) приводило к гибели животных на 2-13 сутки после введения без выраженных клинических проявлений интоксикации. Вскрытие трупов погибших животных и тщательное патологоанатомическое исследование не позволили определить причину смерти мышей.
Таким образом, по результатам эксперимента было заключено, что однократное внутривенное введение фармацевтической композиции мышам в дозах от 4,3×1011 ф.ч./м2 до 430×1011 ф.ч./м2 удовлетворительно переносится и может быть рекомендовано в указанных пределах для внутривенного введения человеку.
Пример 6
Определение пролонгированности действия.
Изучение фармакокинетики является одним из основных аспектов доклинического исследования лекарственных препаратов, поскольку позволяет обоснованно разрабатывать режимы применения препаратов в клинике.
Фармакокинетику фармацевтической композиции оценивали по экспрессии рекомбинантного лактоферрина методом иммуноферментного анализа с определением стандартных фармакокинетических параметров: максимальной концентрации целевого белка в сыворотке крови (Cmax), времени достижения максимальной концентрации в сыворотке крови (tmax), площади под фармакокинетической кривой концентрация-время (AUC) и периоду полувыведения (t1/2). Фармакокинетику изучали после однократного внутривенного введения фармацевтической композиции в дозах 4,3×1011, 4,3×1012 и 4,3×1013 ф.ч./м2. Дозы были выбраны, исходя из максимально переносимых доз (МПД) фармацевтической композиции, определенных в экспериментах по изучению его «острой» токсичности на мышах, где МПД для мышей - 4,3×1013 ф.ч./м2, при введении которой наблюдалась максимальная продукция рекомбинантного лактоферрина человека в сыворотке крови экспериментальных животных.
В качестве препарата сравнения использовалилактоферрин человека, выделенный из женского молока, который вводили однократно внутривенно в дозе 10 мг/кг.
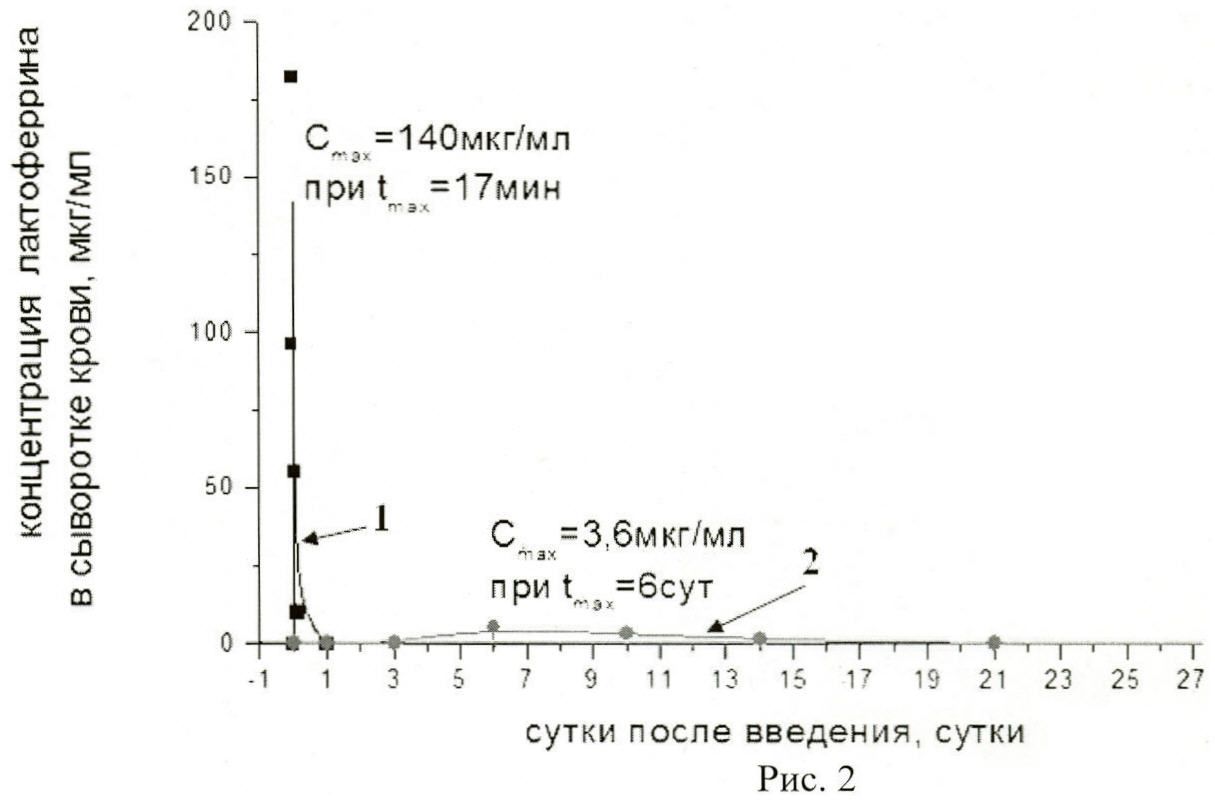
На рисунке 2 представлены:
фармакокинетическая кривая 1  характеризует концентрацию в крови нативного лактоферрина человека из донорского женского молока;
характеризует концентрацию в крови нативного лактоферрина человека из донорского женского молока;
фармакокинетическая кривая 2  - концентрацию в крови рекомбинантного лактоферрина человека, полученную после введения фармацевтической композиции мышам в дозе 4,3×1011 ф.ч./м2;
- концентрацию в крови рекомбинантного лактоферрина человека, полученную после введения фармацевтической композиции мышам в дозе 4,3×1011 ф.ч./м2;
Cmax - максимальная концентрация лактоферрина в сыворотке крови;
tmax - время достижения максимальной концентрации лактоферрина в сыворотке крови.
Таким образом, показано, что концентрация нативного лактоферрина человека (кривая 1) достигала максимального уровня - 140±32 мкг/мл через 17 мин (0,011 суток) после введения, период полувыведения составлял t1/2 часа. При однократном введении фармацевтической композиции в дозе 4,3×1011 ф.ч./м2 Cmax лактоферрина человека (кривая 2) составляла 3,6±0,5 мкг/мл и достигала максимального уровня на 6-е сутки. Эта концентрация белка поддерживалась в сыворотке крови в течение 3-х дней, что говорит о динамическом равновесии между продукцией целевого белка и его выведением. Видно, что две кривые имеют разную форму, различную величину максимума и неодинаковое время достижения максимальной концентрации, но площади под этими кривыми близки по величине, и следовательно, оба лекарственных средства обеспечивают поступление в кровь одинакового количества лекарственного вещества. Время полувыведения (t1/2) лактоферрина человека при введении фармацевтической композиции составляет 8,4 суток, что превышает в 105 раз таковое при введении препарата нативного лактоферрина (t1/2=0,08 суток) и, соответственно, подтверждает увеличение времени присутствия лактоферрина человека в кровотоке.
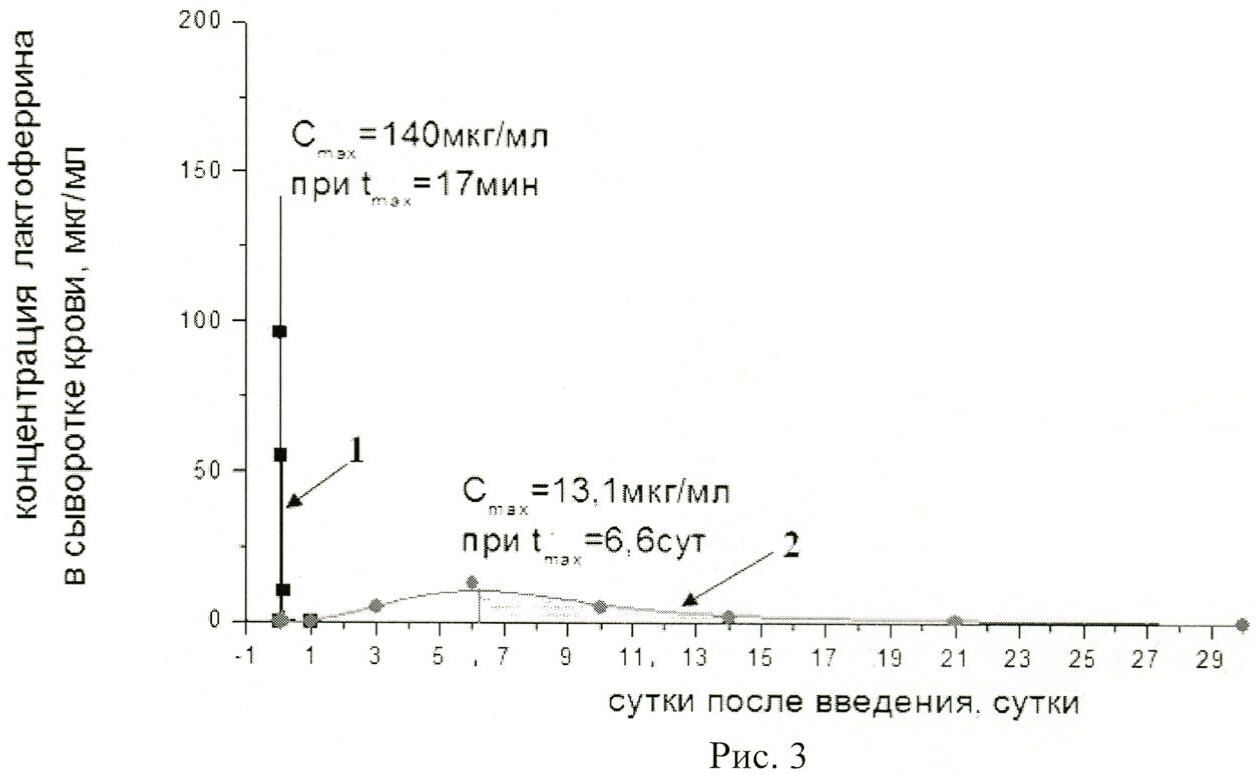
На рисунке 3 представлены:
фармакокинетическая кривая 1 характеризует концентрацию в крови нативного лактоферрина человека из донорского женского молока;
фармакокинетическая кривая 2 - концентрацию в крови рекомбинантного лактоферрина человека, полученную, после введения фармацевтической композиции мышам в дозе 4,3×1012 ф.ч./м2;
Cmax - максимальная концентрация лактоферрина в сыворотке крови;
tmax - время достижения максимальной концентрации лактоферрина в сыворотке крови.
При внутривенном введении фармацевтической композиции в дозе 4,3×1012 ф.ч./м2 Cmax повышается примерно в 4 раза относительно введения фармацевтической композиции в дозе 4,3×1011 ф.ч./м2 и несмотря на то, что она остается ниже, чем при введении нативного лактоферрина, площадь под кривой 2 (фармацевтическая композиция) в 5 раз больше, чем площадь под кривой 1, за счет непрерывной продукции рекомбинантного белка в течение длительного времени.
Введение фармацевтической композиции в дозе 4,3×1013 ф.ч./м2 позволяет достоверно оценить поступление нереплицирующихся наночастиц (по продукции целевого белка) в органы и выведения из них. Поэтому фармакокиненику оценивали и при введении фармацевтической композиции в дозе 4,3×1013 ф.ч./м2.
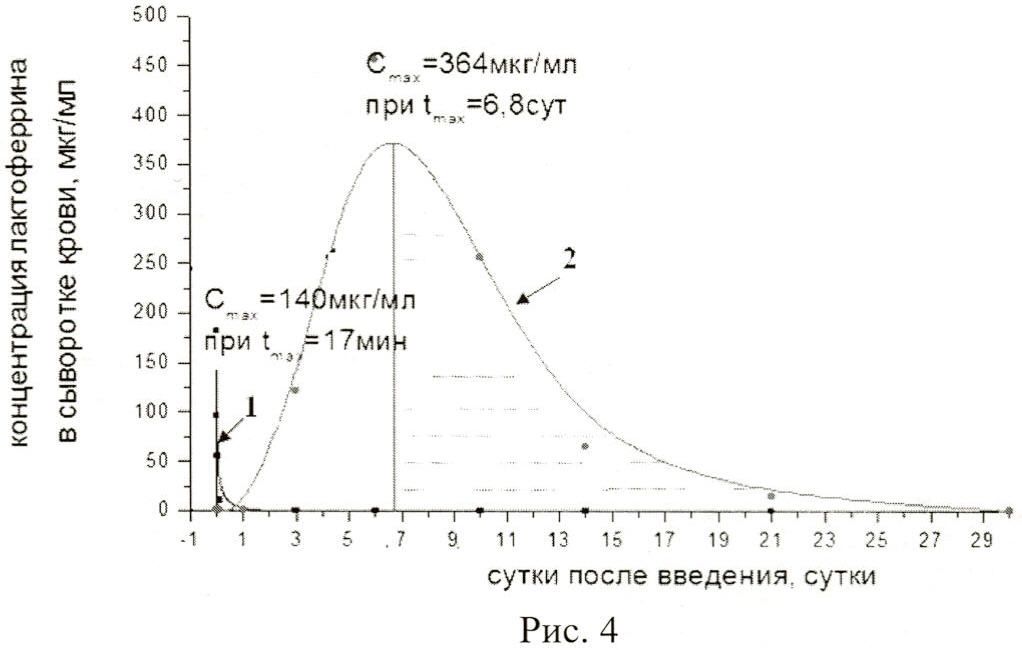
На рисунке 4 представлены:
фармакокинетическая кривая 1 характеризует концентрацию в крови нативного лактоферрина человека из донорского женского молока;
фармакокинетическая кривая 2 - концентрацию в крови рекомбинантного лактоферрина человека, полученную после введения фармацевтической композиции мышам в дозе 4,3×1012 ф.ч./м2;
Cmax - максимальная концентрация лактоферрина в сыворотке крови;
tmax - время достижения максимальной концентрации лактоферрина в сыворотке крови.
После внутривенного введения фармацевтической композиции в дозе 4,3×1013 ф.ч./м2 через 6,8 суток в сыворотке крови достигалась максимальная концентрация рекомбинантного лактоферрина человека, равная 364 мкг/мл, с последующим двухфазным снижением концентрации продуцируемого лактоферрина человека в сыворотке крови. На рисунке видно, что первая фаза биораспределения продолжалась 9,4 суток, при которой период полувыведения (t1/2) составлял 5,4 суток, вторая фаза - 13,8 суток с t1/2=4,8 суток. Площадь под фармакокинетической кривой 2 (фармацевтическая композиция) в 73 раза больше, чем под кривой 1.
Таким образом, сравнение общей концентрации рекомбинантного лактоферрина человека, присутствующего в организме животных при введении различных доз фармацевтической композиции позволяет заключить, что доза 4,3×1011 ф.ч./м2 продуцирует рекомбинантного лактоферрина человека примерно в количестве, соответствующем однократному внутривенному введению нативного лактоферрина в дозе 10 мг/кг, превышение дозы композиции в 10 раз (4,3×1012 ф.ч./м2) приводит к увеличению продукции лактоферрина в 3,6 раз, а введение фармацевтической композиции в дозе 4,3×1013 ф.ч./м2 - в 101 раз. Фармакокинетические кривые отражают пролонгированное действие фармацевтической композиции с 12 часов до 30 суток после введения.
Пример 7
Определение пути введения фармацевтической композиции.
Оценку местнораздражающего действия фармацевтической композиции при ее однократном внутривенном введении осуществляли макроскопически и с использованием микроскопических (гистологических) методов исследования.
Исследование проводили на 12 кроликах породы «Шиншилла», самцах. Фармацевтическую композицию вводили кроликам однократно внутривенно (в краевую вену уха) в объеме 1,0 мл в дозе 4,3×1011 ф.ч./м2. Контрольным животным внутривенно вводили изотонический (0,9%) раствор хлористого натрия в том же режиме.
Материал (фрагмент уха кролика с краевой веной) для микроскопического (гистологического) исследования отбирали на 3-й и 14-е сутки после введения фармацевтической композиции.
Фрагменты уха с веной фиксировали в 10% нейтральном формалине, после чего от каждого образца брали 2 поперечно срезанных участка уха с веной и подвергали последующей общепринятой гистологической обработке, включавшей промывку в проточной воде, обезвоживание в спиртах, пропитку в хлороформе и парафине, заливку в парафин, резку парафиновых блоков на микротоме. Срезы толщиной 5 мкм после депарафинизации окрашивали гематоксилином и эозином, заключали в канадский бальзам. Гистологические препараты исследовали в световом микроскопе серии МС 300 фирмы Micros (Австрия) при увеличениях 100, 400, 1000.
Критериями оценки местнораздражающего действия являлись:
- макроскопические изменения: внешние изменения со стороны сосуда (уплотнение сосуда, гиперемия окружающей кожи и воспалительная реакция в области прохождения сосуда);
- микроскопические критерии: патологические изменения стенки вены, развитие тромбофлебита, образование тромбов.
Визуальный (макроскопический) анализ места введения фармацевтической композиции показал, что у кроликов в месте введения препарата (краевая вена уха) в течение всего срока наблюдения отсутствовали: уплотнение сосуда, гиперемия окружающей кожи и воспалительная реакция в области прохождения сосуда. Таким образом, макроскопически, признаков, свидетельствующих о местнораздражающем действии фармацевтической композиции при ее однократном внутривенном введении не выявлено.
При гистологическом исследовании мест введения фармацевтической композиции наблюдали следующую картину:
1) у контрольных кроликов на 3 и 14 сутки после внутривенного введения 0,9% раствора хлористого натрия у всех кроликов просвет вены был несколько расширен, свободен, с небольшим содержанием крови. Патологические изменения стенки вены и окружающих подкожных тканей не обнаружены.
2) у кроликов, получивших фармацевтическую композицию, на 3 сутки после однократного внутривенного введения фармацевтической композиции вены у всех кроликов были расширены с небольшим содержанием крови. У всех животных отмечено некоторое выбухание кожи над сосудом. При гистологическом исследовании у 2-х кроликов патологических изменений стенки вены и окружающих тканей не обнаружено. У одного кролика отмечен небольшой отек - выход плазмы в стенку вены и окружающую подкожную ткань.
На 14 сутки после внутривенного введения фармацевтической композиции у всех кроликов просвет вены был не расширен и содержал небольшое количество крови. Признаков отека или иных патологических изменений в стенке вены и окружающих тканей не выявлено.
Таким образом, однократное внутривенное введение фармацевтической композиции оказывало слабо выраженное и полностью обратимое местнораздражающее действие. Противопоказаний для внутривенного применения фармацевтической композиции не выявлено.
Пример 8.
Определение необходимости разведения для реализации способа введения.
Так как в ходе предыдущих исследований (пример 4) был рекомендован внутривенный путь введения фармацевтической композиции, проводили экспериментальную оценку ее совместимости с кровью. Для этого оценивали гемолитический потенциал фармацевтической композиции и формулирующего буфера.
Фармацевтическую композицию и формулирующий буфер разводили 0,9% раствором хлористого натрия. Пробы инкубировали в термостате при температуре 37°C.
|
Представленные в таблице 6 данные показывают, что в течение 1-го часа гемолиз эритроцитов отсутствовал (процент гемолиза эритроцитов в опытных и контрольной (инкубация крови с 0,9% раствором хлористого натрия) пробах составил 3,4±0,9% и 2,2±0,2%, соответственно. При дальнейшей инкубации от 2-х до 24-х часов наблюдали постепенное увеличение гемолиза эритроцитов в опытных пробах с 4,5% до 100%. В то же время гемолиз эритроцитов в контрольных пробах не превысил величину в 2,8%. так при введении неразведенного формулирующего буфера начиная с 30 минут после экспозиции, время полного гемолиза наступило в обоих случаях через 24 часа. Гемолиз эритроцитов в контрольном веществе (0,9% растворе хлористого натрия). При оценке гемолитического потенциала стабилизирующего буферного раствора были получены аналогичные результаты. Так, при инкубации буферного раствора с кровью в течение 1-го часа гемолиз эритроцитов в опытных пробах отсутствовал, а при увеличении времени инкубации от 2 до 24 часов отмечали нарастание гемолиза эритроцитов в пробах от 5,8±0,7% до 100%.
Таким образом, данные, полученные в модельной системе, свидетельствуют о наличии у фармацевтической композиции гемолитической активности. При этом гемолитическая активность фармацевтической композиции обусловлена гемолитической активностью формулирующего буфера.
Было заключено, что разведение раствора в 80 и 800 раз 0,9% раствором хлористого натрия позволило снизить гемолитическую активность препарата до контрольных величин (по 0,9% раствору хлористого натрия).
Пример 9.
Определение приемлемого растворителя
Для внутривенного введения фармацевтической композиции в разведении в 80 раз (пример 8) человеку исследовали пригодные для внутривенного введения растворы - вода для инъекций, 5% раствор глюкозы, 10% раствор глюкозы, 0,9% раствор хлористого натрия. Оценивали стабильность содержащихся в фармацевтической композиции нереплицирующихся наночастиц при разведении каждым из этих растворов.
Для постановки опыта 3 мл композиции с содержанием 2,33×1011 ф.ч./мл разводили в 200 мл растворителя, получали разведение в 80 раз, что соответствовало 2,9×106 ф.ч./мл. Полученную смесь после определенного времени экспозиции при комнатной температуре, вносили на пермиссивную культуру клеток, инкубировали при 37°C и 5% CO2 7 дней. Стабильность содержащихся в композиции нереплицирующихся наночастиц со вставкой гена лактоферрина человека оценивали по изменению их титров (оценивали в единицах активности по наличию бляшкообразования).
|
Оценка результатов, представленных в таблице 7, показала сохранение стабильности нереплицирующихся наночастиц без значимой потери титров в 5% и 10% растворах глюкозы, также как и в контрольном веществе. Однако в 0,9% растворе хлористого натрия и воде для инъекций титры нереплицирующихся наночастиц упали до нуля, что означает отсутствие стабильности нереплицирующихся наночастиц в них.
Таким образом, полученные результаты позволяют рекомендовать в качестве приемлемого растворителя для растворения и внутривенного введения фармакологической композиции 5% и 10% растворы глюкозы.
Пример 10.
Подтверждение безопасности способа введения разработанной композиции
Данные, характеризующие влияние фармацевтической композиции, растворенной в 5% и 10% растворах глюкозы на свертываемость крови крыс ex vivo, представлены в таблице 8. Для постановки опыта 3 мл композиции с содержанием 2,33×1011 ф.ч./мл разводили в 200 мл растворителя, получали разведение в 80 раз, что соответствовало 2,9×108 ф.ч./мл. В исследовании использовали нестабилизированную кровь крыс. Пробы инкубировали в термостате при температуре 37°C. Пробы оценивали визуально при пристальном наблюдении за образцами в течение 3-х минут.
|
Результаты, представленные в таблице 8 показывают, что фармацевтическая композиция, разведенная 5% или 10% раствором глюкозы хорошо смешивается с кровью и не приводят к образованию сгустков. Время свертывания крови опытных проб, а также 0,9% раствора хлористого натрия (контрольное вещество) было сопоставимым и составляло 427,33±11,56; 392,67±27,09 и 361,67±9,28 секунд, соответственно. Время свертывания нативной крови составляло 368,00±15,31 секунд.
Таким образом, при внесении фармацевтической композиции в указанных концентрациях и формулирующего буфера в цельную нестабилизированную венозную кровь крыс изменений в пробах, которые свидетельствовали бы о несовместимости данной фармацевтической композиции и буферного раствора с кровью не выявлено.
Пример 11
Определение способа введения фармацевтической композиции человеку.
Предыдущие экспериментальные исследования
фармацевтической композиции проводили на мышах, у которых не бывает предсуществующего иммунного ответа к аденовирусу человека. Однако известно, что многие люди могут иметь предсуществующий иммунный ответ к аденовирусам человека вследствие естественного инфицирования. Мощный иммунный ответ быстро нейтрализует вводимые конструкции, что может значительно снизить терапевтический эффект препарата. (Harvey B.-G., Hackett N.R., El-Sawy Т. et al. Variability of human systemic humoral responses to adenovirus gene transfer vectors administrated to different organs. J. Virol, 1999, v.73, pp.6729-6742 - Разнообразие гуморального ответа у человека на введение аденовирусных векторов в различные органы - стр.6729).
Эффективность заявленной схемы введения фармацевтической композиции в отношении снятия предсуществующего иммунного ответа была установлена в экспериментах на мышах с искусственно индуцированным предсуществующим иммунным ответом к аденовирусу человека 5 серотипа (Ad5).
Для получения модели предсуществующего иммунного ответа мыши линии Balb/c весом 7-9 грамм трехкратно внутримышечно иммунизировали аденовирусом человека 5-го серотипа, не несущим в себе вставку гена лактоферрина, в дозе 3×1011 ф.ч. на мышь. Через три недели после иммунизации в сыворотках крови мышей в реакции нейтрализации определяли уровень вирус-нейтрализующих антител к аденовирусу человека пятого серотипа. Титр антител к аденовирусу человека при внутримышечном введении мышам составил 1:64 и был отличен от контрольной группы (1:8 - неспецифические титры).
Заявленная схема введения фармацевтической композиции основана на особенностях иммунной системы, позволяющих после первичного введения специфического антигена и связывания его с «предсуществующими» антителами некоторое время не образовывать новые антитела в ответ на повторную стимуляцию специфическим антигеном. Эффективность заявленной схемы введения фармацевтической композиции подтвердили экспериментально. Первой группе мышей с индуцированным предсуществующим иммунным ответом к аденовирусу человека 5-го серотипа вводили однократно внутривенно рекомендованную в предыдущих исследованиях по специфической активности полную дозу фармацевтической композиции - 4.3×1011 ф.ч./м2, позволяющую достичь максимальной концентрации лактоферрина в крови (Cmax=3,6 мкг/мл) на 6-е сутки после введения. Второй группе мышей с предсуществующим иммунным ответом к аденовирусу 5-го серотипа полную дозу композиции (4.3×1011 ф.ч./м2) вводили дробно внутривенно, в первый день вводили одну треть (1,43×1011 ф.ч./м2), через сутки ввели полную дозу фармацевтической композиции (4.3×1011 ф.ч./м2). Для контроля сформировали еще две группы из мышей, не имеющих предсуществующго иммунного ответа к аденовирусу человека 5 серотипа, одной группе вводили фармацевтичекую композицию, содержащую дозу 4.3×1011 ф.ч./м2, другой - эквивалентный объем физиологического раствора NaCl. Все опытные и контрольные группы состояли из 5 животных.
Для регистрации полученного эффекта по снятию предсуществующего иммунного ответа у всех мышей взяли сыворотки крови на 6-е сутки и определяли концентрацию лактоферрина человека в них (таблица 9).
|
Результаты эксперимента, представленные в таблице 9 показали, что внутривенное введение одной трети от полной дозы фармацевтической композиции (1,43×1011 ф.ч./м2) эффективно снимает предсуществующеий иммунный ответ к аденовирусу человека 5-го серотипа. Это позволило при внутривенном введении полной дозы композиции через сутки, составляющей 4,3×1011 ф.ч./м2, добиться экспрессии лактоферрина человека в терапевтической концентрации (3,58±0,51 мкг/кг) на 6-е сутки после введения фармацевтической композиции, что сопоставимо с концентрацией рекомбинантного лактоферрина, полученной после однократного введения композиции в полной дозе мышам, не имеющим предсуществующего иммунного ответа. У мышей с предсуществующим иммунным ответом введение композиции однократно вполной дозе не позволило получить высокую концентрацию рекомбинантного лактоферрина в сыворотке крови (0,8±0,12 мкг/кг).
Таким образом, схема введения фармацевтической композиции, основанная на предварительном внутривенном введении одной трети основной дозы (1,43×1011 ф.ч./м2) и последующем внутривенном введении полной дозы композиции через сутки (4,3×1011 ф.ч./м2), позволяет получить терапевтическую концентрацию рекомбинантного лактоферрина в организме, имеющем предсуществующий иммунный ответ к аденовирусу человека пятого серотипа.
Далее в клинических примерах, дозы, полученные во время доклинических исследований и измеряемые на м2 поверхности тела были переведены на человека, т.к. являются эквивалентными (средняя площадь поверхности тела человека равна 1,62 м2). (Хабриев Р.У., Руководство по экспериментальному (доклиническому) изучению новых фармакологических веществ, 2000 г., 98 стр.) (Guidance for Industry. Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers - Руководство для промышленности. Оценка максимальной безопасной стартовой дозы в начальных клинических испытаниях для терапии у взрослых здоровых добровольцев. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER).July 2005, Pharmacology and Toxicology - 7, 19 стр.)
Пример 12
Определение способа терапии человеку Больной Л. Рак щитовидной железы IV ст. II кл. гр. (T4N2aM1). Назначено химиотерапевтическое лечение: циклофосфан, винкристин, доксорубицин. В начале лечения возникли тяжелые нарушения со стороны системы крови - эритропения, лимфопения, а также общая слабость, тошнота, диспепсия. После внутривенного капельного введение фармацевтической композиции в 67 мл 10% раствора глюкозы в дозе 2,33·×1011 ф.ч. и последующем внутривенном капельном введении полной дозы фармацевтической композиции в 200 мл 10% раствора глюкозы через сутки (7×1011 ф.ч.) были улучшены гематологические показатели, а также устранены тошнота и диспепсия, общее состояние больного улучшилось. Далее проводили профилактическое введение фармацевтической композиции по той же схеме за 24 часа до начала следующего курса химиотерапии.
Пример 13
Определение способа терапии человеку
Больной В. Лимфосаркома IV ст. (генерализованная форма), состояние после хирургического лечения. Вследствие проведения дальнейшего химиотерапевтического лечения (циклофосфан) возникло умеренное токсическое поражение печени, пневмония, эритро- и лимфопения. Для устранения указанных токсических эффектов применяли внутривенное капельное введение фармацевтической композиции в 67 мл 5% раствора глюкозы в дозе 2,33×1011 ф.ч. и последующее внутривенное капельное введение полной дозы фармацевтической композиции в 200 мл 5% раствора глюкозы через сутки (7×1011 ф.ч.). Применение фармацевтической композиции по данной схеме способствовало купированию пневмонии, поражение печени и улучшило гемодинамические показатели крови.
Пример 14
Определение способа терапии человеку.
Больная П. Метахромный рак правой молочной железы T3N0M0. состояние после мастэктомии. В начале проведения курса постоперационной лучевой терапии (разовая доза 2 Гр ежедневно на постоперационный рубец) возникла местная лучевая реакция в виде воспалительной реакции кожи в области облучения, а также больная испытывала реакции общего характера - нарушение функции желудочно-кишечного тракта (снижение аппетита, тошнота, рвота, диарея). Для устранения указанных токсических эффектов применяли внутривенное капельное введение фармацевтической композиции в 67 мл 10% раствора глюкозы в дозе 2,33×1011 ф.ч. и последующее внутривенное капельное введение полной дозы фармацевтической композиции в 200 мл 10% раствора глюкозы через сутки (7×1011 ф.ч.). Применение фармацевтической композиции по данной схеме позволило улучшить общее состояние больной, а также устранить воспалительные реакции в месте облучения. Показатели ACT, АЛТ, креатинина и мочевины в норме.
В результате, заявленная фармацевтическая композиция содержит в своем составе нереплицирующиеся наночастицы на основе генома аденовируса человека 5-го серотипа со вставкой гена лактоферрина человека, позволяющая при введении в дозах от 7×1011 ф.ч. до 7×1013 ф.ч. на человека, достичь пролонгированной (от 12 часов до 29 дней) экспрессии лактоферрина человека. Экспрессирующийся фармацевтической композицией лактоферрин человека соответствует нативному лактоферрину из женского донорского молока по физико-химическим свойствам, антиоксидантной активности и антимикробным свойствам. Детоксицирующее действие фармацевтической композиции достигается при введении в дозах от 7×1011 ф.ч. до 7×1012 ф.ч. на человека. Для осуществления заявленного способа терапии предложена фармацевтическая композиция, которая две формы выпуска - 1 мл и 3 мл, с содержанием нереплицирующихся наночастиц в дозе не менее 2,33×1011 ф.ч./мл. По заявленному способу терапии в первый раз фармацевтическая композиция вводится человеку внутривенно капельно в дозе 2,33×1011 ф.ч., содержащейся в 1 мл композиции, которую предварительно разбавляют 67 мл 5% или 10% раствора глюкозы. Второе введение композиции осуществляют через сутки - полную дозу 7×1011 ф.ч., содержащуюся в 3 мл композиции, предварительно разбавляют 200 мл 5% или 10% раствора глюкозы и вводят внутривенно капельно. Заявленный способ терапии позволяет безопасно для организма человека и терапевтически эффективно лечить токсикозы различной этиологии у людей, имеющих предшествующий иммунный ответ к аденовирусу человека 5-го серотипа, что говорит о достижении задач, поставленных в данном изобретении.
Таким образом, по сравнению с прототипом, преимуществами заявленной фармацевтической композиции на основе нереплицирующихся наночастиц, продуцирующей антиоксидантный белок человека - лактоферрин являются:
- однократное введение для получения терапевтической концентрации в организме человека;
- пролонгированное действие (до 3-х недель);
- снижение затрат препаратов, медицинского инструментария, рабочего времени медицинского персонала;
- отпадение необходимости использования женского молока для получения лактоферрина;
- снижение стоимости производства;
- возможность получения сравнительно большего количества лактоферрина.
Таким образом, поставленная техническая задача, направленная на создание фармацевтической композиции на основе нереплицирующихся наночастиц, продуцирующей непосредственно в организме антиоксидантный белок человека-лактоферрин, обладающий антиоксидантным, антимикробным, антитоксическим белком, пригодной для лечения токсических состояний различной этиологии и обеспечивающей достижение стойкого терапевтического эффекта при однократных введениях этой композиции выполнена. При использовании заявленной фармацевтической композиции обеспечивается сокращение затрат препарата, медицинского инструментария и времени медицинского персонала для достижения требуемого результата лечения.
Использование в фармацевтической и клинической практике заявляемой фармацевтической композиции и способов ее применения позволяет достичь нескольких технических, лечебных и экономических результатов:
- заявляемая фармацевтическая композиция биосовместима с организмом человека и терапевтически высоко эффективна;
- нереплицирующиеся наночастицы, продуцирующие антиоксидантные белки человека совместимы с различными носителями - растворителями для внутривенного использования;
- фармацевтическая композиция пригодна для лечения заболеваний различной локализации;
Способ оценки риска прогрессирования немелкоклеточного рака легкого после хирургического лечения
Способ прогнозирования послеоперационных осложнений у больных с опухолевым поражением легкого
Фотосенсибилизаторы для фотодинамической терапии
Фотосенсибилизатор для фотодинамической терапии
Рекомбинантная трехвалентная вакцина от гриппа человека
Фармацевтическая композиция для терапии острых токсических состояний
Фармацевтическая композиция и способ терапии нейродегенеративных заболеваний, в частности бокового амиотрофического склероза
Рекомбинантная псевдоаденовирусная частица на основе генома аденовируса человека 5 серотипа для индукции специфического иммунитета к вирусу гриппа а субтипа н1n1 и способ ее использования в качестве компонента для создания вакцины
Рекомбинантная псевдоаденовирусная частица на основе генома аденовируса человека 5 серотипа, для индукции специфического иммунитета к вирусу гриппа а субтипа н3n2 и способ ее использования
Рекомбинантная псевдоаденовирусная частица на основе генома аденовируса человека 5 серотипа, продуцирующая гемагглютинин вируса гриппа штамма b/brisbane/60/2008, способ ее использования для индукции специфического иммунитета к вирусу гриппа в
Способ оценки риска прогрессирования немелкоклеточного рака легкого после хирургического лечения
Способ прогнозирования послеоперационных осложнений у больных с опухолевым поражением легкого
Фотосенсибилизаторы для фотодинамической терапии
Фотосенсибилизатор для фотодинамической терапии
Рекомбинантная трехвалентная вакцина от гриппа человека
Фармацевтическая композиция для терапии острых токсических состояний
Фармацевтическая композиция и способ терапии нейродегенеративных заболеваний, в частности бокового амиотрофического склероза
Рекомбинантная псевдоаденовирусная частица на основе генома аденовируса человека 5 серотипа для индукции специфического иммунитета к вирусу гриппа а субтипа н1n1 и способ ее использования в качестве компонента для создания вакцины
Рекомбинантная псевдоаденовирусная частица на основе генома аденовируса человека 5 серотипа, для индукции специфического иммунитета к вирусу гриппа а субтипа н3n2 и способ ее использования
Рекомбинантная псевдоаденовирусная частица на основе генома аденовируса человека 5 серотипа, продуцирующая гемагглютинин вируса гриппа штамма b/brisbane/60/2008, способ ее использования для индукции специфического иммунитета к вирусу гриппа в

