Результат интеллектуальной деятельности: ДИПЕПТИДНЫЕ ПРОЛЕКАРСТВА И ИХ ПРИМЕНЕНИЕ
Вид РИД
Изобретение
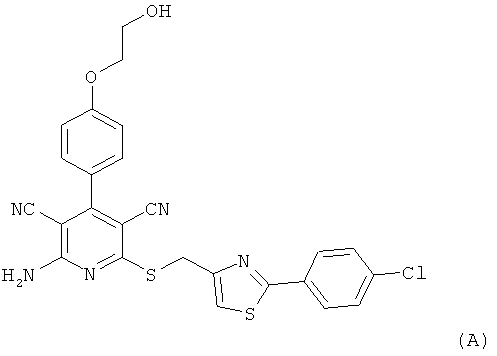
Настоящее изобретение относится к пролекарствам, которые являются производными 2-амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}тио)-4-[4-(2-гидроксиэтокси)фенил]пиридин-3,5-дикарбонитрила, способу их получения, их применению для лечения и/или профилактики болезней, а также к их применению для изготовления лекарственных средств, предназначенных для лечения и/или профилактики болезней, прежде всего сердечно-сосудистых заболеваний.
Пролекарства являются производными действующего вещества, которые перед высвобождением собственно действующего вещества претерпевают происходящее in vivo одноступенчатое или многоступенчатое биопревращение ферментативного и/или химического характера. Пролекарства, как правило, содержат химическую группировку, способствующую улучшению комплекса свойств лежащего в их основе действующего вещества [Р.Ettmayer и другие, J. Med. Chem., 47, 2393 (2004)]. При этом для обеспечения оптимального фармацевтического действия структуру указанной химической группировки и предусматриваемый механизм высвобождения действующего вещества из пролекарства следует приводить в чрезвычайно точное соответствие с индивидуальным действующим веществом, показанием для его применения, местом его действия и способом применения. Многие лекарственные средства применяют в виде пролекарств, которые в отличие от лежащих в их основе действующих веществ характеризуются более высокой биодоступностью, что достигается, например, благодаря оптимизации физико-химических характеристик подобных пролекарств, в частности повышению растворимости, а также улучшению способности к активному и пассивному поглощению или специфичному для тех или иных тканей распределению. Из обширного перечня касающейся пролекарств литературы особо следует упомянуть, например, Н.Bundgaard (издатель), Design of Prodrugs: Bioreversible derivatives for various functional groups and chemical entities, издательство Elsevier Science Publishers B.V., 1985.
Аденозин (пуриновый нуклеозид) присутствует в любых клетках и выделяется в свободном виде в качестве одного из многих физиологических и патофизиологических стимулирующих веществ. Аденозин образуется в качестве промежуточного продукта при деструкции аденозин-5'-монофосфата (AMP) и S-аденозилгомоцистеина внутри клеток, однако может высвобождаться из них и вследствие присоединения к специфическим рецепторам оказывать воздействие в качестве гормонального вещества или нейротрансмиттера. Посредством аденозин 1-рецепторов оказывают воздействие на эссенциальные функции прежде всего в возбудимых и/или действующих клетках различных тканей [смотри, например, K.A.Jacobson, Z.-G.Gao, Nat. Rev. Drug Discover., 5, 247-264 (2006)].
2-Амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}тио)-4-[4-(2-гидрокси-этокси)фенил]пиридин-3,5-дикарбонитрил [соединение формулы (А)] является эффективным при оральном применении агонистом рецептора аденозина 1, находящимся в настоящее время на стадии углубленного клинического исследования в качестве потенциального нового фармацевтического действующего вещества, предназначенного прежде всего для профилактики и лечения сердечно-сосудистых заболеваний [смотри WHO Drug Information, том 20, 2 (2006), а также получение и применение указанного соединения согласно примеру 6 международной заявки WO 03/053441].
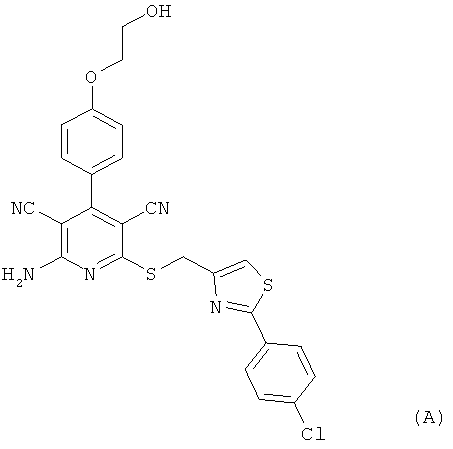
Однако соединение формулы (А) обладает низкой растворимостью в воде, физиологических средах и органических растворителях, а также ограниченной биодоступностью при оральном применении суспензий этого кристаллического вещества. Указанные обстоятельства, с одной стороны, обусловливают ограничение внутривенного применения соединения формулы (А) в качестве действующего вещества чрезвычайно низкими дозировками, поскольку приготовление инфузионных растворов на основе физиологических солевых растворов с использованием обычных гидротропных солюбилизаторов оказывается весьма затруднительным. С другой стороны, затруднительным является также изготовление соответствующих лекарственных препаратов в виде таблеток. Исходя из вышеизложенного в основу настоящего изобретения была положена задача предложить производные или пролекарства соединения формулы (А), которые обладают повышенной растворимостью в указанных выше средах и/или улучшенной биодоступностью при оральном применении и вместе с тем после применения способны контролируемо высвобождаться в организме пациента. Повышенная пригодность соединения формулы (А) для внутривенного применения могла бы открыть другие сферы его терапевтического применения в качестве действующего вещества.
Обзор пролекарств, производных сложных эфиров карбоновых кислот, и возможные свойства этих соединений опубликованы, например, в K.Beaumont и другие в Curr. Drug Metab., 4, 461-485 (2003).
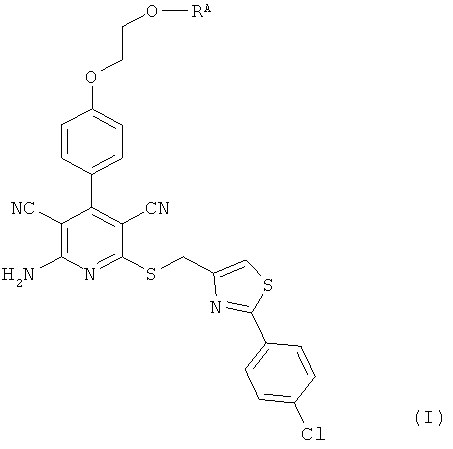
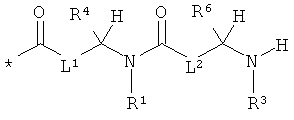
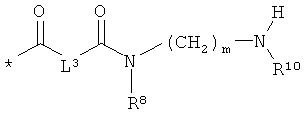
Объектом настоящего изобретения являются соединения общей формулы (I):
в которой
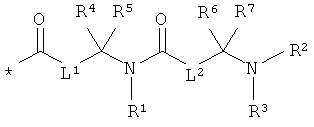
RA означает группу формулы:
 ,
, 
или 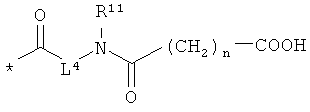
в которой
* означает место присоединения к атому кислорода,
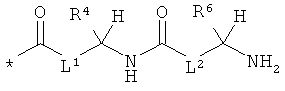
L1 и L2 независимо друг от друга означают связь или -CH2-группу,
R1, R2 и R3 независимо друг от друга означают водород или метил,
R4 и R6 одинаковые или разные и независимо друг от друга означают водород или боковую группу природной α-аминокислоты или ее гомологов или изомеров,
R5 и R7 независимо друг от друга означают водород или метил,
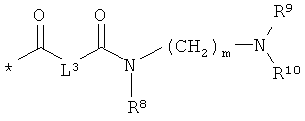
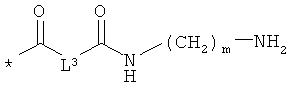
L3 означает неразветвленный или разветвленный алкандиил с 2-4 атомами углерода, который замещен аминогруппой,
R8, R9 и R10 независимо друг от друга означают водород или метил,
m означает 2, 3, 4, 5 или 6,
L4 означает неразветвленный или разветвленный алкандиил с 2-4 атомами углерода, который замещен карбоксильной группой,
R11 означает водород или метил, и
n означает 1, 2, 3 или 4,
а также их соли, сольваты и сольваты солей.
Предлагаемыми в изобретении соединениями являются соединения формулы (I), их соли, сольваты и сольваты солей, указанные ниже другие соединения формулы (I) и их соли, сольваты и сольваты солей, а также указанные в приведенных ниже примерах осуществления изобретения другие соединения формулы (I), их соли, сольваты и сольваты солей, если под указанными ниже другими соединениями формулы (I) уже не подразумеваются соли, сольваты и сольваты солей.
Предлагаемые в изобретении соединения в зависимости от их структуры могут находиться в форме стереоизомеров (энантиомеров или диастереомеров). Таким образом, настоящее изобретение относится также к энантиомерам или диастереомерам, а также к их смесям. Индивидуальные стереоизомеры могут быть выделены из подобных смесей энантиомеров и/или диастереомеров известными методами.
В случае, если предлагаемые в изобретении соединения находятся в виде таутомеров, изобретение относится ко всем без исключения таутомерным формам подобных соединений.
В соответствии с настоящим изобретением предпочтительными солями являются физиологически приемлемые соли предлагаемых в изобретении соединений. Под подобными солями подразумевают также соли, которые непригодны для фармацевтического применения, однако могут быть использованы, например, для выделения или очистки предлагаемых в изобретении соединений. Помимо солей, содержащих один солеобразующий остаток, согласно настоящему изобретению при необходимости используют также возможные соли, содержащие несколько, в частности два или три, солеобразующих остатков.
Физиологически приемлемыми солями предлагаемых в изобретении соединений являются образующиеся по реакции присоединения соли минеральных кислот, карбоновых кислот и сульфокислот, например соли соляной кислоты, бромистоводородной кислоты, серной кислоты, фосфорной кислоты, метансульфокислоты, этансульфокислоты, толуолсульфокислоты, бензолсульфокислоты, нафталиндисульфокислоты, уксусной кислоты, трифторуксусной кислоты, пропионовой кислоты, молочной кислоты, винной кислоты, яблочной кислоты, лимонной кислоты, фумаровой кислоты, малеиновой кислоты или бензойной кислоты.
Физиологически приемлемыми солями предлагаемых в изобретении соединений являются также соли обычных оснований, например предпочтительно соли щелочных металлов (в частности, соли натрия или калия), соли щелочноземельных металлов (в частности, соли кальция или магния) и соли аммония, которые являются производными аммиака или органических аминов с 1-16 атомами углерода, например предпочтительно этиламина, диэтиламина, триэтиламина, этилдиизопропиламина, моноэтаноламина, диэтаноламина, триэтаноламина, холина, дициклогексиламина, диметиламиноэтанола, прокаина, дибензиламина, морфолина, N-метилморфолина, аргинина, лизина, этилендиамина, пиперидина и N-метилпиперидина.
В соответствии с настоящим изобретением под сольватами подразумевают такие формы предлагаемых в изобретении соединений, которые представляют собой комплексы, образуемые этими находящимися в твердом или жидком состоянии соединениями вследствие координации молекул растворителя. Гидраты являются особой формой сольватов и возникают вследствие координации указанными соединениями молекул воды. В соответствии с настоящим изобретением предпочтительными сольватами являются гидраты.
В соответствии с настоящим изобретением в отсутствие особых указаний заместители означают следующее.
Алкандиилом с 2-4 атомами углерод в соответствии с настоящим изобретением является неразветвленный или разветвленный двухвалентный алкильный остаток с 2-4 атомами углерода. Предпочтительным является неразветвленный алкандиильный остаток с 2-4 атомами углерода. Примерами предпочтительных алкандиилов являются этан-1,2-диил (1,2-этилен), этан-1,1-диил, пропан-1,3-диил (1,3-пропилен), пропан-1,1-диил, пропан-1,2-диил, пропан-2,2-диил, бутан-1,4-диил (1,4-бутилен), бутан-1,2-диил, бутан-1,3-диил и бутан-2,3-диил. Кроме того, алкандиильный остаток в случае группы L3 замещен аминогруппой, а в случае группы L4 карбоксильной группой.
Под боковыми группами α-аминокислот, используемыми в качестве заместителей R4 и R6, подразумевают боковые группы природных α-аминокислот, а также их гомологов или изомеров. Соответствующие α-аминокислоты могут обладать как L-, так и D-конфигурацией, или могут представлять собой смесь α-аминокислот L- и D-конфигураций. Примерами пригодных боковых групп являются метил (аланин), пропан-2-ил (валин), пропан-1-ил (норвалин), 2-метилпропан-1-ил (лейцин), 1-метилпропан-1-ил (изолейцин), бутан-1-ил (норлейцин), трет-бутил (2-трет-бутилглицин), фенил (2-фенилглицин), бензил (фенилаланин), п-гидроксибензил (тирозин), индол-3-илметил (триптофан), имидазол-4-илметил (гистидин), гидроксиметил (серии), 2-гидроксиэтил (гомосерин), 1-гидроксиэтил (треонин), меркаптометил (цистеин), метилтиометил (S-метилцистеин), 2-меркаптоэтил (гомоцистеин), 2-метилтиоэтил (метионин), карбамоилметил (аспарагин), 2-карбамоилэтил (глутамин), карбоксиметил (аспарагиновая кислота), 2-карбоксиэтил (глутаминовая кислота), 4-аминобутан-1-ил (лизин), 4-амино-3-гидроксибутан-1-ил (гидроксилизин), 3-аминопропан-1-ил (орнитин), 2-аминоэтил (2,4-диаминомасляная кислота), аминометил (2,3-диаминопропионовая кислота), 3-гуанидинопропан-1-ил (аргинин) и 3-уреидопропан-1-ил (цитрулин). Под боковыми группами α-аминокислот, используемыми в качестве заместителя R4, предпочтительно подразумевают метил (аланин), пропан-2-ил (валин), пропан-1-ил (норвалин), 2-метилпропан-1-ил (лейцин), 1-метилпропан-1-ил (изолейцин), бутан-1-ил (норлейцин), бензил (фенилаланин), п-гидроксибензил (тирозин), имидазол-4-илметил (гистидин), гидроксиметил (серин), 1-гидроксиэтил (треонин), карбамоилметил (аспарагин) и 2-карбамоилэтил (глутамин). Под боковыми группами α-аминокислот, используемыми в качестве заместителя R6, предпочтительно подразумевают имидазол-4-илметил (гистидин), 4-аминобутан-1-ил (лизин), 3-аминопропан-1-ил (орнитин), 2-аминоэтил (2,4-диаминомасляную кислоту), аминометил (2,3-диаминопропионовую кислоту) и 3-гуанидинопропан-1-ил (аргинин). Предпочтительными являются соответствующие α-аминокислоты, которые обладают L-конфигурацией.
Предпочтительными являются соединения формулы (I), в которой:
RA означает группу формулы:
 ,
, 
или 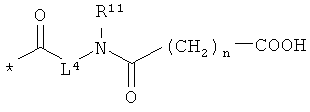 ,
,
в которой
* означает место присоединения к атому кислорода,
L1 означает связь,
L2 означает связь или -CH2-группу,
R1 и R3 независимо друг от друга означают водород или метил,
R4 означает водород, метил, пропан-2-ил, пропан-1-ил, 2-метилпропан-1-ил, 1-метилпропан-1-ил, бутан-1-ил, бензил, п-гидроксибензил, имидазол-4-илметил, гидроксиметил, 1-гидроксиэтил, карбамоилметил или 2-карбамоилэтил,
R6 означает водород, имидазол-4-илметил, 4-аминобутан-1-ил, 3-аминопропан-1-ил, 2-аминоэтил, аминометил или 3-гуанидинопропан-1-ил,
L3 означает неразветвленный алкандиил с 2-4 атомами углерода, который замещен аминогруппой,
R8 и R10 независимо друг от друга означают водород или метил,
m означает 2, 3 или 4,
L4 означает неразветвленный алкандиил с 2-4 атомами углерода, который замещен карбоксильной группой,
R11 означает водород или метил, и
n означает 2, 3 или 4,
а также их соли, сольваты и сольваты солей.
Особенно предпочтительными являются соединения формулы (I), в которой
RA означает группу формулы:
 ,
, 
или 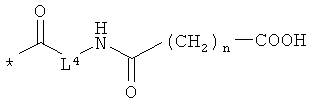
в которой
* означает место присоединения к атому кислорода,
L1 и L2 соответственно означают связь,
R4 означает водород, метил, пропан-2-ил, пропан-1-ил, 2-метилпропан-1-ил, 1-метилпропан-1-ил, бутан-1-ил, бензил, п-гидроксибензил, имидазол-4-илметил, гидроксиметил, 1-гидроксиэтил, карбамоилметил или 2-карбамоилэтил,
R6 означает имидазол-4-илметил, 4-аминобутан-1-ил, 3-аминопропан-1-ил, 2-аминоэтил, аминометил или 3-гуанущинопропан-1-ил,
L3 означает группу формулы -CH(NH2)-CH2-, -CH2-CH(NH2)-, -CH2-CH(NH2)-CH2-, -CH(NH2)-CH2-CH2- или -CH2-CH2-CH(NH2)-,
m означает 2, 3 или 4,
L4 означает группу формулы **-CH2-СН(СООН)- или **-CH2-CH2-СН(СООН)-, в которой
** означает место присоединения к соседней карбонильной группе,
и
n означает 2 или 3,
а также их соли, сольваты и сольваты солей.
Еще более предпочтительными являются соединения формулы (I), в которой:
RA означает группу формулы:
в которой
* означает место присоединения к атому кислорода,
L1 и L2 соответственно означают связь,
R4 означает водород, метил, пропан-2-ил, 2-метилпропан-1-ил, бензил, гидроксиметил или 1-гидроксиэтил, и
R6 означает имидазол-4-илметил, 4-аминобутан-1-ил, 3-аминопропан-1-ил, 2-аминоэтил, аминометил или 3-гуанидинопропан-1-ил,
а также их соли, сольваты и сольваты солей.
Еще более предпочтительными являются также соединения формулы (I), в которой:
RA означает группу формулы:
в которой
* означает место присоединения к атому кислорода,
L3 означает группу формулы -CH(NH2)-CH2-, -CH2-CH(NH2)-, -CH(NH2)-CH2-CH2- или -CH2-CH2-CH(NH2)-, и
m означает 2 или 3,
а также их соли, сольваты и сольваты солей.
Другим объектом настоящего изобретения является способ получения предлагаемых в изобретении соединений формулы (I), отличающийся тем, что соединение формулы (А):
[А] в инертном растворителе в присутствии средства конденсации этерифицируют карбоновой кислотой формулы (II), (III) или (IV):
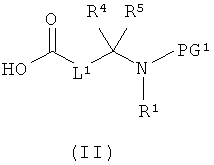
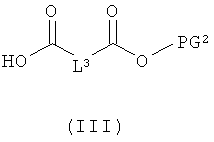
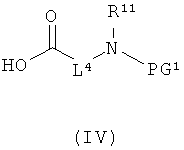
в которой
L1, L3, L4, R1, R4, R5 и R11 соответственно такие, как указано выше,
PG1 означает временную аминозащитную группу, например, такую как трет-бутоксикарбонил, и
PG2 означает временную карбоксилзащитную группу, например, такую как трет-бутил,
получая соединения формулы (V), (VI), соответственно (VII):
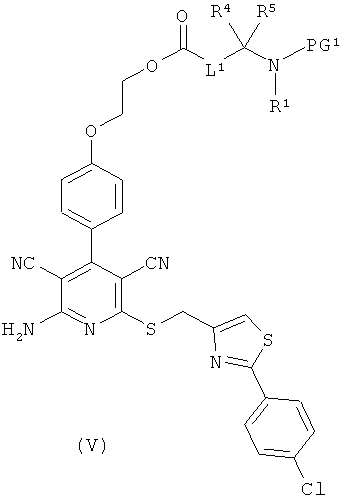
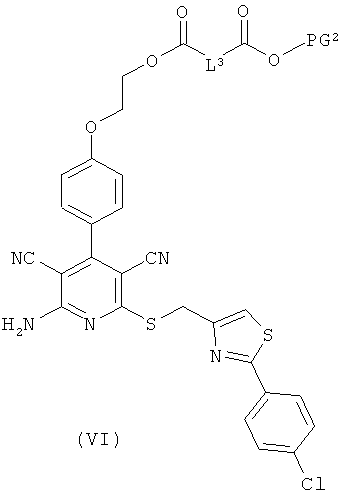
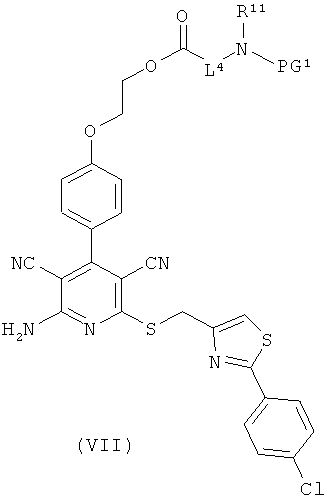
в которой L1, L3, L4, R1, R4, R5, R11, PG1 и PG2 соответственно такие, как указано выше,
затем отщепляют защитную группу PG1, соответственно PG2, и в инертном растворителе в присутствии средства конденсации сочетают в случае соединения (V) с соединением формулы (VIII), в случае соединения (VI) с соединением формулы (IX) и в случае соединения (VII) с соединением формулы (X):
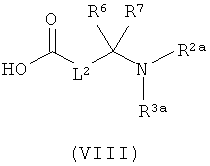
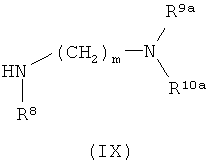
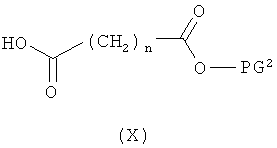
в которых
L2, R6, R7, R8, PG2, m и n соответственно такие, как указано выше, и
R2a и R3a, а также R9a и R10a, соответственно одинаковые или разные и такие, как указано выше для R2, R3, R9, соответственно R10, или означают временную аминозащитную группу, например, такую как трет-бутоксикарбонил,
после чего удаляют при необходимости присутствующие защитные группы,
или
[В] в инертном растворителе в присутствии средства конденсации сочетают с соединением формулы (XI), (XII) или (XIII):
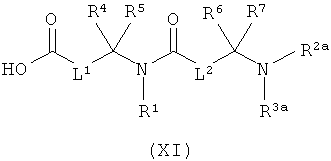
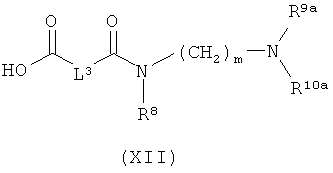
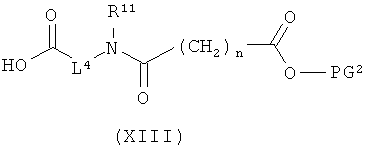
в которой
L1, L2, L3, L4, R1, R4, R5, R6, R7, R8, R11, m и n соответственно такие, как указано выше,
R2a и R3a, а также R9a и R10a, соответственно одинаковые или разные и такие, как указано выше для R2, R3, R9, соответственно R10, или означают временную аминозащитную группу, например, такую как трет-бутоксикарбонил, и
PG2 означает временную карбоксилзащитную группу, например, такую как трет-бутил,
после чего удаляют при необходимости присутствующие защитные группы,
и полученные соединения формулы (I) с использованием соответствующих (i) растворителей и/или (ii) кислот или оснований при необходимости переводят в их сольваты, соли и/или сольваты солей.
Таким образом, превращение (А)→(I) в соответствии с вариантом [А] выполняют путем последовательного сочетания отдельных, при необходимости надлежащим образом защищенных компонентов (карбоновой кислоты, соответственно амина) или в соответствии с вариантом [В] путем непосредственного ацилирования надлежащим образом защищенного дипептидного производного. При этом реакцию сочетания (образования сложного эфира, соответственно амида) реализуют известными из химии пептидов методами [смотри, например, М.Bodanszky, Principles of Peptide Synthesis, издательство Springer, Берлин, 1993; H.-D.Jakubke, H.Jeschkeit, Aminosauren, Peptide, Proteine, издательство Chemie, Вейнгейм, 1982].
В качестве инертных растворителей для осуществления реакций сочетания используют, например, простые эфиры, такие как диэтиловый эфир, трет-бутилметиловый эфир, диоксан, тетрагидрофуран, диметиловый эфир этиленгликоля или диметиловый эфир диэтиленгликоля, углеводороды, такие как бензол, толуол, ксилол, гексан, циклогексан или нефтяные фракции, галогенированные углерводороды, такие как дихлорметан, трихлорметан, тетрахлорметан, 1,2-дихлорэтан, трихлорэтилен или хлорбензол, или другие растворители, такие как ацетон, этилацетат, пиридин, диметилсульфоксид, диметилформамид, N,N'-диметилпропиленмочевина, N-метилпирролидон или ацетонитрил. Наряду с этим можно использовать смеси указанных растворителей. Предпочтительными растворителями являются дихлорметан, диметилформамид или их смеси.
В качестве средств конденсации для осуществления реакций сочетания используют, например, карбодиимиды, такие как N,N'-диэтилкарбодиимид, N,N'-дипропилкарбодиимид, N,N'-диизопропилкарбодиимид, N,N'-дициклогексилкарбодиимид или N-(3-диметиламиноизопропил)-N'-этилкарбодиимид гидрохлорид, производные фосгена, такие как N,N'-карбонилдиимидазол, соединения 1,2-оксазолия, такие как 2-этил-5-фенил-1,2-оксазолий-3-сульфат или перхлорат 2-трет-бутил-5-метилизоксазолия, ациламиносоединения, такие как 2-этокси-1-этоксикарбонил-1,2-дигидрохинолин, или изобутилхлорформиат, ангидрид пропанфосфоновой кислоты, диэтиловый эфир цианфосфоновой кислоты, бис(2-оксо-3-оксазолидинил)фосфорилхлорид, гексафторфосфат бензотриазол-1-илокситрис-(диметиламино)фосфония, гексафторфосфат бензотриазол-1-илокситрис-(пирролидино)фосфония, тетрафторборат O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония, гексафторфосфат O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония, тетрафторборат 2-(2-оксо-1-(2Н)-пиридил)-1,1,3,3-тетраметилурония, гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония или тетрафторборат O-(1Н-6-хлорбензотриазол-1-ил)-1,1,3,3-тетраметилурония, при необходимости в комбинации с другими вспомогательными веществами, такими как 1-гидроксибензотриазол или N-гидроксисукцинимид, а также используемыми в качестве оснований карбонатами щелочных металлов, например карбонатом натрия или карбонатом калия, или органическими основаниями, такими как триэтиламин, N-метилморфолин, N-метилпиперидин, N,N-диизопропилэтиламин или 4-N,N-диметиламинопиридин. Для образования эфира предпочтительно используют N-(3-диметиламиноизопропил)-N'-этилкарбодиимид гидрохлорида в комбинации с 4-N,N-диметиламинопиридином. Для образования амида предпочтительно используют N-(3-диметиламиноизопропил)-N'-этилкарбодиимид гидрохлорида в комбинации с 1-гидроксибензотриазолом или N-гидроксисукцинимидом и при необходимости основанием, таким как N,N-диизопропилэтиламин.
Реакции сочетания в общем случае выполняют в температурном интервале от 0 до +60°С, предпочтительно от +20 до +40°С. Их можно осуществлять при нормальном, повышенном или пониженном давлении (например, от 0,5 до 5 бар). Реакции сочетания в общем случае осуществляют при нормальном давлении.
Соединения формулы (I) могут быть получены также указанными выше способами непосредственно в виде солей. Подобные соли путем выполняемой в инертном растворителе обработки основанием, соответственно кислотой, хроматографическими методами или посредством ионообменных смол при необходимости могут быть преобразованы в соответствующие свободные основания, соответственно кислоты. Другие соли предлагаемых в изобретении соединений при необходимости могут быть получены также путем замены противоионов посредством ионообменной хроматографии, например, предусматривающей использование смол Amberlite®.
При реализации описанных выше реакционных последовательностей при необходимости присутствующие в остатках R4, R6, L3 и/или L4 функциональные группы, прежде всего аминогруппы, гуанидиногруппы, гидроксильные группы, меркаптогруппы и карбоксильные группы, в случае целесообразности или необходимости могут находиться также во временно защищенной форме. При этом введение и удаление соответствующих защитных групп осуществляют обычными, известными из химии пептидов методами [смотри, например, T.W.Greene, P.G.M.Wuts, Protective Groups в Organic Synthesis, издательство Wiley, Нью-Йорк, 1999; M.Bodanszky, A.Bodanszky, The Practice of Peptide Synthesis, издательство Springer, Берлин, 1984].
В качестве групп, предназначенных для защиты амино и гуанидино, предпочтительно используют трет-бутоксикарбонил (Boc) или бензилоксикарбонил (Z). В качестве групп, предназначенных для защиты гидроксильных или карбоксильных функциональных групп, предпочтительно используют трет-бутил или бензил. Указанные защитные группы отщепляют обычными методами, предпочтительно путем реализуемого в инертном растворителе, таком как диоксан, дихлорметан или уксусная кислота, взаимодействия защищенного соединения с сильной кислотой, такой как водородхлорид, водородбромид или трифторуксусная кислота; отщепление при необходимости можно выполнять также без дополнительного использования инертного растворителя. В случае если в качестве защитной группы используют бензил и бензилоксикарбонил, она может быть удалена также гидрогенолизом в присутствии палладиевого катализатора. Отщепление указанных защитных групп при необходимости можно выполнять одновременно или на отдельных реакционных стадиях.
Соединения формул (II), (III), (IV), (VIII), (IX), (X), (XI), (XII) и (XIII) коммерчески доступны, известны из литературы или могут быть получены известными из литературы методами. Так, например, соединения формул (II) и (VIII), в которых L1, соответственно L2, означает CH2-группу, могут быть получены из соответствующих соединений, в которых L1, соответственно L2, означает связь, известными методами удлинения цепей карбоновых кислот, например, по реакции Арндта-Эйстерта [Eistert и другие, Ber. Dtsch. Chem. Ges., 60, 1364-1370 (1927); Ye и другие, Chem. Rev., 94, 1091-1160 (1994); Cesar и другие, Tetrahedron Lett., 42, 7099-7102 (2001)] или взаимодействием с N-гидрокси-2-тиопиридином [смотри Barton и другие, Tetrahedron Lett., 32, 3309-3312 (1991)].
Получение соединения формулы (А) (2-амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]-метил}тио)-4-[4-(2-гидроксиэтокси)фенил]пиридин-3,5-дикарбонитрила) описано в примере 6 международной заявки WO 03/053441.
Получение предлагаемых в изобретении соединений представлено на приведенных ниже схемах (расшифровка сокращений приведена ниже).
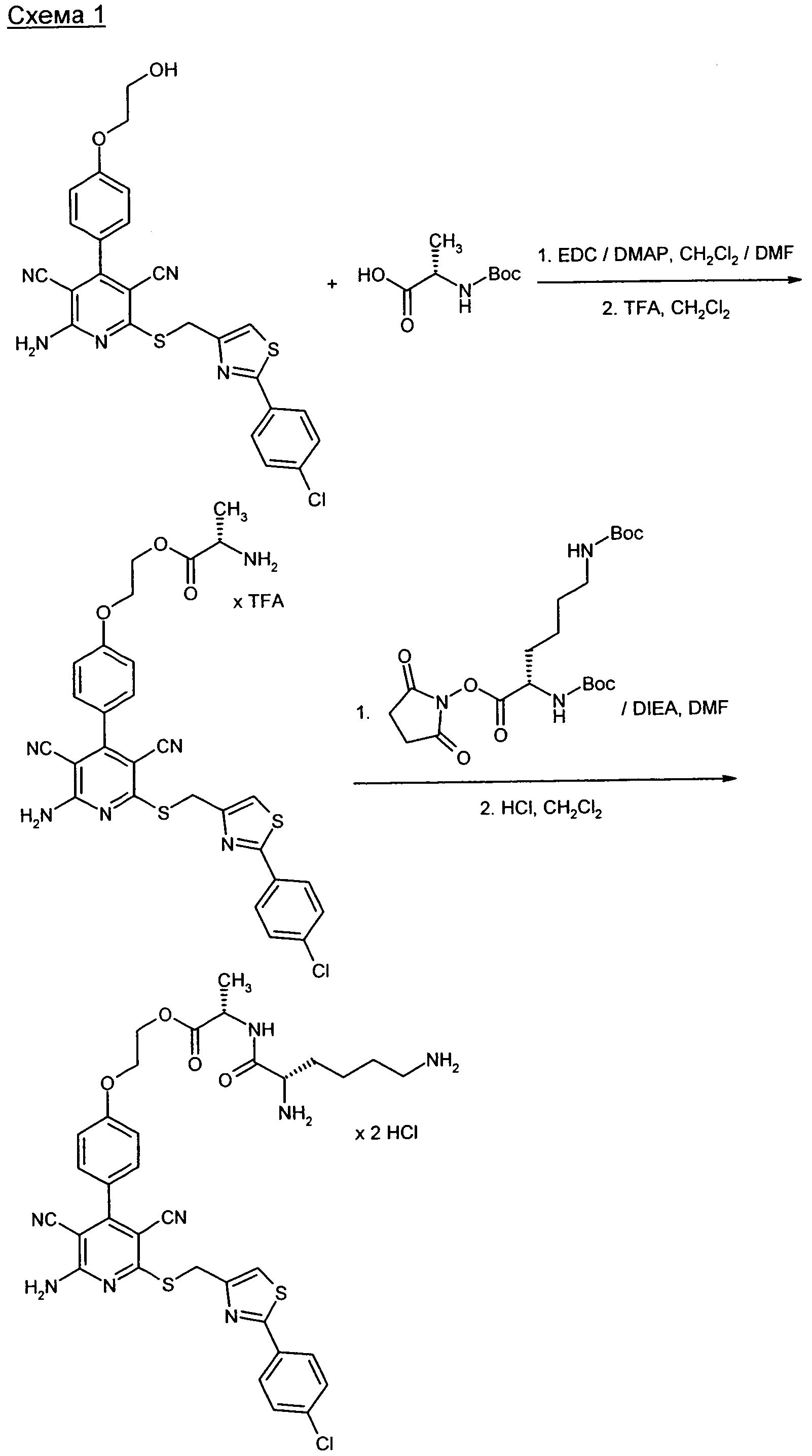
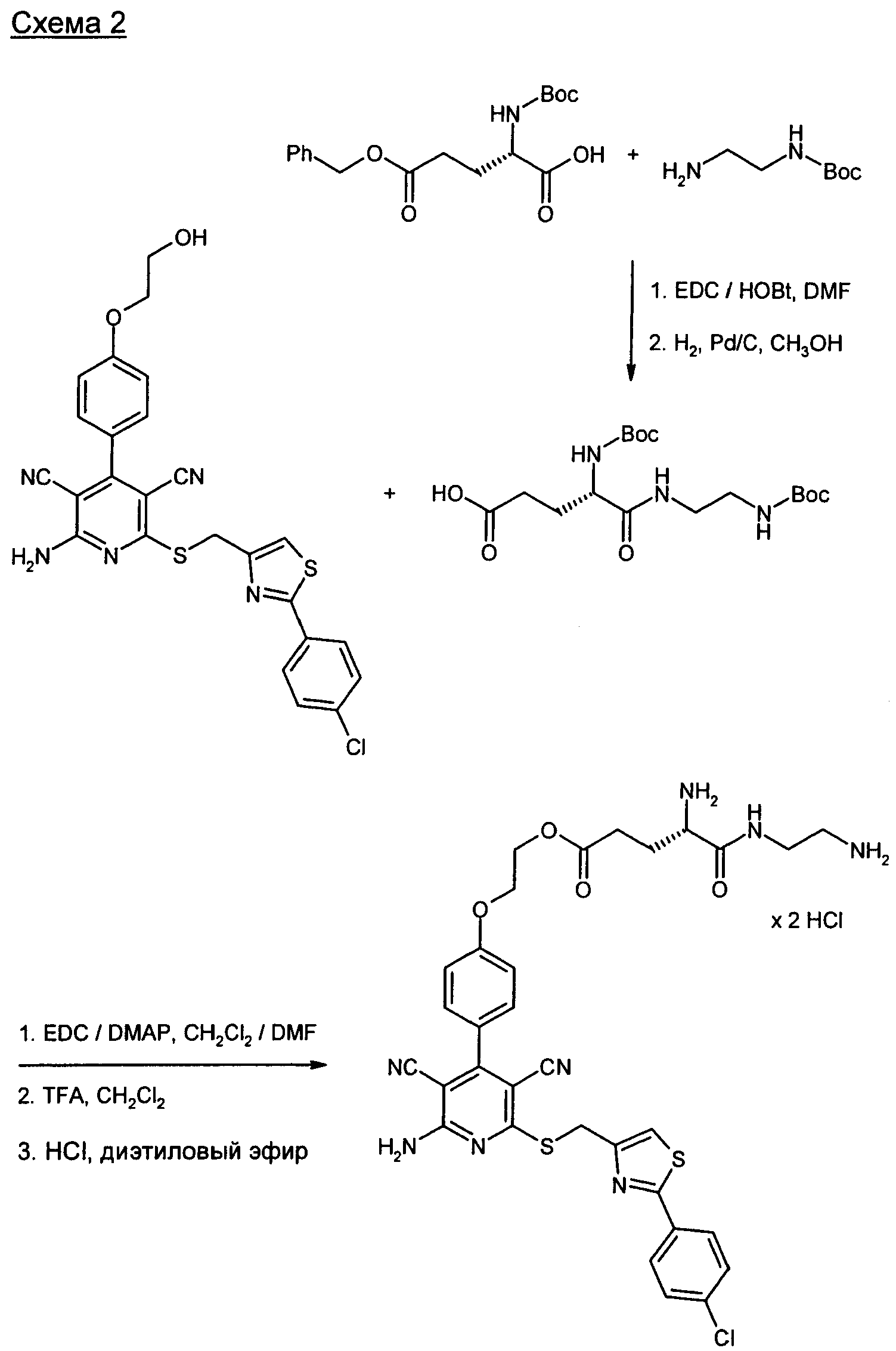
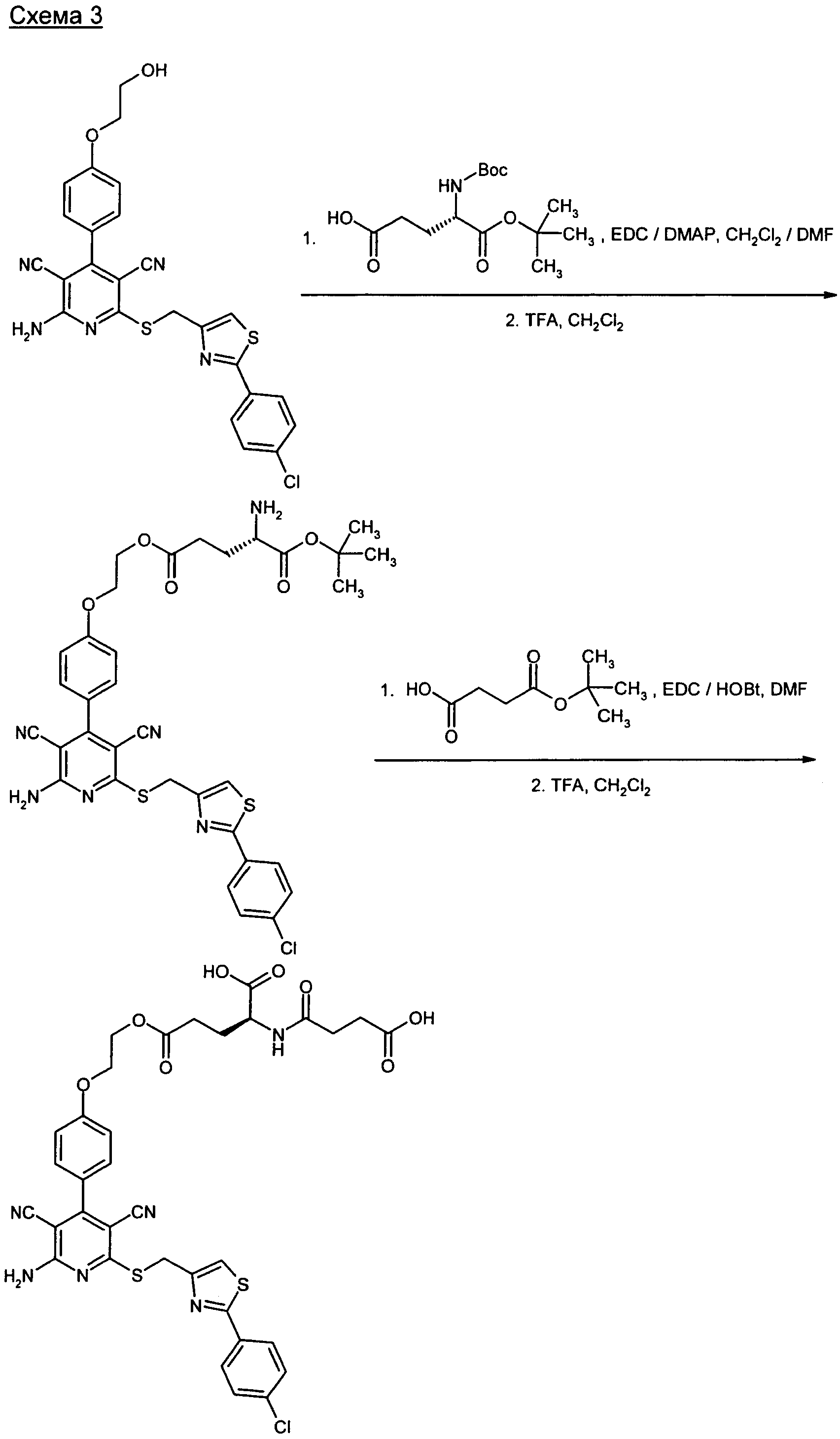
Предлагаемые в изобретении соединения и их соли являются оптимальными пролекарствами соединения формулы (А) в качестве действующего вещества. Они обладают высокой стабильностью, например, при различных значениях показателя pH и в то же время претерпевают эффективное превращение в соединение формулы (А) при физиологическом показателе pH и прежде всего in vivo. Помимо этого предлагаемые в изобретении соединения обладают высокой растворимостью в воде или других физиологически совместимых средах, что обусловливает их пригодность для терапевтического, прежде всего внутривенного применения. Кроме того, при их оральном применении наблюдается повышенная биодоступность по сравнению с исходным соединением формулы (А).
Соединения формулы (I), одни или в комбинации с одним или несколькими другими действующие веществами, пригодны для профилактики и/или лечения различных заболеваний, прежде всего, например, заболеваний сердечно-сосудистой системы, для кардиозащиты после поражений сердца, а также обменных заболеваний.
В соответствии с настоящим изобретением под заболеваниями сердечнососудистой системы подразумевают, например, гипертонию (повышенное кровяное давление), периферические и кардиальные сосудистые заболевания, ишемическую болезнь сердца, коронарные рестенозы, например рестенозы после баллонной дилатации периферических кровеносных сосудов, инфаркт миокарда, острый коронарный синдром, острый коронарный синдром с подъемом сегмента ST, острый коронарный синдром без подъема сегмента ST, стабильную и нестабильную стенокардию, сердечную слабость, стенокардию Принцметала, персистирующую ишемическую дисфункцию («гибернацию миокарда»), преходящую постишемическую дисфункцию («оглушенный миокард»), сердечную недостаточность, тахикардию, предсердную тахикардию, аритмии, мерцания предсердий, мерцания желудочков, персистирующие мерцания предсердий, постоянные мерцания предсердий, мерцания предсердий с нормальной функцией левого желудочка, мерцания предсердий с ограничением функции левого желудочка, синдром преждевременного возбуждения желудочков, нарушения периферического кровообращения, повышенный уровень фибриногена и липопротеинов низкой плотности, а также повышенную концентрацию ингибитора 1 активатора плазминогена (РАI-1), прежде всего гипертонию, ишемическую болезнь сердца, острый коронарный синдром, стенокардию, сердечную недостаточность, инфаркт миокарда и мерцания предсердий.
В соответствии с настоящим изобретением термин «сердечная недостаточность» используют для обозначения как острых, так и хронических форм проявления сердечной недостаточности, а также специфических или схожих картин этого заболевания, таких как острая декомпенсированная сердечная недостаточность, недостаточность правого желудочка сердца, недостаточность левого желудочка сердца, глобальная недостаточность, ишемическая кардиомиопатия, дилатационная кардиомиопатия, врожденный порок сердца, клапанный порок сердца, сердечная недостаточность при клапанном пороке, сужение митрального клапана, недостаточность митрального клапана, стеноз клапана аорты, недостаточность клапана аорты, сужение трехстворчатого клапана, недостаточность трехстворчатого клапана, стеноз клапана легочной артерии, недостаточность клапана легочной артерии, комбинированный клапанный порок сердца, воспаление сердечной мышцы (миокардит), хронический миокардит, острый миокардит, вирусный миокардит, диабетическая сердечная недостаточность, кардиомиопатия, вызванная алкоголизмом, сердечный тезаурисмоз, а также диастолическая и систолическая сердечная недостаточность.
Кроме того, предлагаемые в изобретении соединения пригодны также прежде всего для восстановления пораженной вследствие инфаркта области миокарда, а также для профилактики повторных инфарктов.
Наряду с этим предлагаемые в изобретении соединения пригодны прежде всего для профилактики и/или лечения тромбоэмболических заболеваний, реперфузионных поражений после ишемии, микрососудистых и макрососудистых поражений (васкулита), артериальных и венозных тромбозов, отеков, ишемий, таких как инфаркт миокарда, кровоизлияния в мозг и транзиторных ишемических приступов, для защиты сердца при аортокоронарном шунтировании, первичной чрескожной транслюминальной коронарной ангиопластике, чрескожной транслюминальной коронарной ангиопластике после тромболиза, неотложной чрескожной транслюминальной коронарной ангиопластике, трансплантации сердца и операций на открытом сердце, а также для защиты органов при операциях трансплантации и шунтирования, при обследованиях с использованием катетера и других операционных вмешательствах.
Другие показания для возможного применения предлагаемых в изобретении соединений относятся, например, к профилактике и/или лечению заболеваний урогенитальной сферы, например, таких как острая почечная недостаточность, гиперестезия мочевого пузыря, недержание мочи, эректильная дисфункция и женская сексуальная дисфункция, а также к профилактике и/или лечению воспалительных заболеваний, например, таких как воспалительные дерматозы и артриты, прежде всего ревматические артриты, заболеваний центральной нервной системы и нейродегенеративных нарушений (состояния после инсульта, болезни Альцгеймера, болезни Паркинсона, слабоумия, хореи Гентингтона, эпилепсии, депрессий, множественного склероза), болевых синдромов и мигреней, фиброза печени, онкологических заболеваний, тошноты и рвоты при лечении рака, а также к заживлению ран.
Другие показания для возможного применения предлагаемых в изобретении соединений относятся, например, к профилактике и/или лечению заболеваний дыхательных путей, например, таких как астма, хроническое обструктивное заболевание легких, хронический бронхит, эмфизема легких, бронхоэктазия, кистозный фиброз (муковисцидоз) и пульмональная гипертония, прежде всего пульмональная артериальная гипертония.
Предлагаемые в изобретении соединения можно использовать также для профилактики и/или лечения обменных заболеваний, например, таких как диабет, прежде всего сахарный диабет, диабет беременных, инсулинозависимый диабет и инсулинонезависимый диабет, диабетических осложнений, например, таких как ретинопатия, нефропатия и нейропатия, метаболических заболеваний, например, таких как метаболический синдром, гипергликемия, гиперинсулинемия, резистентность к инсулину, непереносимость глюкозы и ожирение (болезнь Деркума), а также артериосклероз и дислипидемии (гиперхолестеринемия, гипертриглицеридемия, повышенные концентрации триглицеридов в постпрандиальной плазме, гипоальфалипопротеинемия, комбинированные гиперлипидемии), прежде всего для профилактики и/или лечения диабета, метаболического синдрома и дислипидемии.
Другим объектом настоящего изобретения является применение предлагаемых в изобретении соединений для лечения и/или профилактики заболеваний, прежде всего указанных выше заболеваний.
Другим объектом настоящего изобретения является применение предлагаемых в изобретении соединений для изготовления лекарственного средства, предназначенного для лечения и/или профилактики заболеваний, прежде всего указанных выше заболеваний.
Другим объектом настоящего изобретения является способ лечения и/или профилактики заболеваний, прежде всего указанных выше заболеваний, предусматривающий применение эффективного количества по меньшей мере одного из предлагаемых в изобретении соединений.
Предлагаемые в изобретении соединения можно использовать индивидуально или при необходимости в комбинации с другими действующими веществами. Другим объектом настоящего изобретения являются лекарственные средства, предназначенные прежде всего для лечения и/или профилактики указанных выше заболеваний, которые содержат по меньшей мере одно из предлагаемых в изобретении соединений и одно или несколько других действующих веществ.
Предпочтительно используемыми дополнительными действующими веществами являются, например, действующие вещества, изменяющие жировой обмен, антидиабетические средства, средства для снижения кровяного давления, средства, стимулирующие кровоток, и/или антитромботические средства, антиаритмические средства, антиоксиданты, антагонисты рецептора хемокина, ингибиторы р38-киназы, агонисты нейропептидтирозина, агонисты орексина, средства, подавляющие аппетит, ингибиторы ацетилгидролазы фактора активации тромбоцитов, противовоспалительные средства (ингибиторы циклооксигеназы, антагонисты рецептора лейкотриена В4), а также анальгетические средства, например, такие как аспирин.
Объектом настоящего изобретения прежде всего являются комбинации по меньшей мере одного из предлагаемых в изобретении соединений по меньшей мере с одним изменяющим жировой обмен действующим веществом, антидиабетическим средством, действующим веществом для снижения кровяного давления, антиаритмическим средством и/или антитромботическим средством.
Предлагаемые в изобретении соединения предпочтительно могут быть скомбинированы с одним или несколькими из следующих действующих веществ:
- изменяющие жировой обмен действующие вещества, например, предпочтительно выбранные из группы, включающей ингибиторы HMG-CoA-редуктазы, ингибиторы экспрессии HMG-CoA-редуктазы, ингибиторы синтеза сквалена, ингибиторы АСАТ, индукторы рецептора липопротеина низкой плотности, ингибиторы поглощения холестерина, полимерные поглотители желчной кислоты, ингибиторы реабсорбции желчной кислоты, ингибиторы МТР, ингибиторы липазы, активаторы липопротеин-липазы, фибраты, никотиновую кислоту, ингибиторы СЕТР, α-агонисты PPAR, γ-агонисты PPAR и/или δ-агонисты PPAR, модуляторы RXR, модуляторы FXR, модуляторы LXR, тироидные гормоны и/или тироидные имитаторы, ингибиторы АТР-цитратлиазы, антагонисты Lp(a), 1-антагонисты рецептора канабиноида, агонисты рецептора лептина, агонисты рецептора бомбезина, агонисты рецептора гистамина и антиоксиданты/акцепторы радикалов;
- антидиабетические средства, приведенные в Roten Liste 2004/11, глава 12, а также, например, предпочтительно выбранные из группы, включающей сульфонилкарбамиды, бигуаниды, производные меглитинида, ингибиторы глюкозидазы, ингибиторы дипептидил-пептидазы IV (DPP-IV-ингибиторы), оксадиазолидиноны, тиазолидиндионы, агонисты GLP 1-рецептора, антагонисты глюкагона, сенсибилизаторы инсулина, агонисты ССК 1-рецептора, агонисты рецептора лептина, ингибиторы печеночных ферментов, участвующие в стимуляции гликонеогенеза и/или гликогенолиза, модуляторы усвоения глюкозы, а также средства для открытия калиевого канала, например, приведенные в международных заявках WO 97/26265 и WO 99/03861;
- снижающие кровяное давление действующие вещества, например, предпочтительно выбранные из группы, включающей антагонисты кальция, антагонисты ангитензина АII, ингибиторы АСЕ, ингибиторы ренина, антагонисты бета-адреноцептора, антагонисты альфа-адреноцептора, диуретики, антагонисты альдостерона, антагонисты рецептора минералокортикоидного гормона, ингибиторы ЕСЕ, а также ингибиторы вазопептидазы;
- средства, обладающие противосвертывающим действием, например, предпочтительно выбранные из группы, включающей ингибиторы агрегации тромбоцитов и антитромботические вещества;
- антиаритмические средства, прежде всего предназначенные для лечения суправентрикулярной аритмии и тахикардии;
- вещества для профилактики и лечения ишемических и реперфузионных нарушений;
- антагонисты рецептора вазопрессина;
- органические нитраты и доноры NO;
- соединения с позитивным инотропным действием;
- соединения, ингибирующие деструкцию циклического гуанозинмонофосфата (cGMP) и/или циклического аденозинмонофосфата (сАМР), например, такие как ингибиторы фосфодиэстеразы (PDE) 1, 2, 3, 4 и/или 5, прежде всего ингибиторы PDE 5, такие как сильденафил, варденафил и тадалафил, а также ингибиторы PDE 3, такие как мильрининон;
- натрийуретические пептиды, например, такие как предсердные натрийуретические пептиды (ANP, анаритиды), натрийуретические пептиды В-типов или мозговые натрийуретические пептиды (BNP, незиритиды), натрийуретические пептиды С-типов (CNP), а также уродилатин;
- агонисты рецептора простациклина (IP-рецептора), например, такие как илопрост, берапрост и цикапрост;
- кальциевые сенсибилизаторы, например, предпочтительно такие как левосимендан;
- калиевые биологически активные добавки;
- NO- и гемнезависимые активаторы гуанилатциклазы, прежде всего соединения, приведенные в международных заявках W0 01/19355, WO 01/19776, WO 01/19778, WO 01/19780, WO 02/070462 и WO 02/070510;
- NO-независимые, однако гемзависимые стимуляторы гуанилатциклазы, прежде всего соединения, приведенные в международных заявках WO 00/06568, WO 00/06569, WO 02/42301 и WO 03/095451;
- ингибиторы человеческой нейротрофильной эластазы (HNE), например, такие как сивелестат и DX-890 (релтран);
- соединения, ингибирующие каскады трансдукции сигналов, например, такие как ингибиторы тирозинкиназы, прежде всего сорафениб, иматиниб, гефитиниб и эрлотриниб;
- соединения, которые оказывают воздействие на энергетический метаболизм сердца, например, такие как этомоксир, дихлорацетат, ранолазин и триметазидин;
- анальгетические средства; и/или
- вещества для профилактики и лечения тошноты и рвоты.
Под изменяющими жировой обмен действующими веществами предпочтительно подразумевают соединения, выбранные из группы, включающей ингибиторы HMG-CoA-редуктазы, ингибиторы синтеза сквалена, ингибиторы АСАТ, ингибиторы поглощения холестерина, ингибиторы МТР, ингибиторы липазы, тироидные гормоны и/или тироидные имитаторы, агонисты рецептора ниацина, ингибиторы СЕТР, α-агонисты PPAR, γ-агонисты PPAR и/или δ-агонисты PPAR, полимерные поглотители желчной кислоты, ингибиторы реабсорбции желчной кислоты антагонисты Lp(a), 1-антагонисты рецептора канабиноида, антиоксиданты/акцепторы радикалов, а также 1-антагонисты рецептора канабиноида.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с ингибитором HMG-CoA-редуктазы из класса статинов, например, предпочтительно ловастатином, симвастатином, правастатином, флувастатином, аторвастатином, розувастатином, церивастатином или питавастатином.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с ингибитором синтеза сквалена, например, предпочтительно таким как BMS-188494 или ТАК-475.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с ингибитором АСАТ, например, предпочтительно таким как авасимиб, мелинамид, пактимиб, эфлуцимиб или SMP-797.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с ингибитором поглощения холестерина, например, предпочтительно таким как эцетримиб, тиквесид или памаквесид.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с ингибитором МТР, например, предпочтительно таким как имплитапид, BMS-201038, R-103757 или JTT-130.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с ингибитором липазы, например, предпочтительно таким как орлистат.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с тироидными гормонами и/или тироидными имитаторами, например, предпочтительно такими как D-тироксин или 3,5,3'-трииодотиронин (ТЗ).
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с агонистом рецептора ниацина, например, предпочтительно таким как никотиновая кислота, аципимокс, ацифран или радекол.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с ингибитором СЕТР, например, предпочтительно таким как торцетрапиб, JTT-705, BAY 60-5521, BAY 78-7499 или вакцина СЕТР (авант).
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с γ-агонистом PPAR, например, предпочтительно таким как пиоглитазон или розиглитазон.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с δ-агонистом PPAR, например, предпочтительно таким как GW-501516 или BAY 68-5042.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с полимерным поглотителем желчной кислоты, например, предпочтительно таким как холестирамин, колестипол, колесольвам, холестагель или колестимид.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с ингибитором реабсорбции желчной кислоты, например, предпочтительно таким как ингибитор ASBT (соответственно IBAT), например, AZD-7806, S-8921, АК-105, BARI-1741, SC-435 или SC-635.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с антиоксидантом/акцептором радикалов, например, предпочтительно таким как пробукол, AGI-1067, ВО-653 или AEOL-10150.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с 1-антагонистом рецептора канабиноида, например, предпочтительно таким как римонабант или SR-147778.
Под антидиабетическими средствами предпочтительно подразумевают инсулин и производные инсулина, а также орально применяемые гипогликемические действующие вещества. При этом под инсулином и производными инсулина подразумевают инсулины животного, человеческого или биотехнологического происхождения, а также их смеси. Под орально применяемыми гипогликемическими действующими веществами предпочтительно подразумевают сульфонилкарбамиды, бигуаниды, производные меглитинида, ингибиторы глюкозидазы, ингибиторы DPP-IV и γ-агонисты PPAR.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с инсулином.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с сульфонилкарбамидом, например, предпочтительно таким как толбутамид, глибенкламид, глимепирид, глипизид или гликлазид.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с бигуанидом, например, предпочтительно таким как метформин.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с производным меглитинида, например, предпочтительно таким как репаглинид или натеглинид.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с ингибитором глюкозидазы, например, предпочтительно таким как миглитол или акарбоз.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с ингибитором DPP-IV, например, предпочтительно таким как ситаглиптин или вилдаглиптин.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с γ-агонистом PPAR, например, из класса тиазолидиндионов, например, предпочтительно таким как пиоглитазон или розиглитазон.
Под снижающими кровяное давление средствами предпочтительно подразумевают соединения, выбранные из группы, включающей антагонисты кальция, антагонисты ангитензина АII, ингибиторы АСЕ, ингибиторы ренина, антагонисты бета-адреноцептора, антагонисты альфа-адреноцептора и диуретики.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с антагонистом кальция, например, предпочтительно таким как нифедипин, амлодипин, верапамил или дилтизем.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с антагонистом ангитензина АII, например, предпочтительно таким как лосартан, валсартан, кандесартан, эмбусартан, олмесартан или телмисартан.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с ингибитором АСЕ, например, предпочтительно таким как эналаприл, каптоприл, лизиноприл, рамиприл, делаприл, фозиноприл, квиноприл, периндоприл или трандоприл.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с ингибитором ренина, например, предпочтительно таким как алискирен, SPP-600 или SPP-800.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с антагонистом бета-адреноцептора, например, предпочтительно таким как пропранолол, атенолол, тимолол, пиндолол, альпренолол, окспренолол, пенбутолол, бупранолол, метипранолол, надолол, мепиндолол, каразалол, соталол, метопролол, бетаксолол, целипролол, бисопролол, картеолол, эсмолол, лабеталол, карведилол, адапролол, ландиолол, небиволол, эпанолол или буциндолол.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с антагонистом альфа-адреноцептора, например, предпочтительно таким как працозин.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с диуретиком, например, предпочтительно таким как фуросемид, буматанид, торсемид, бендрофлуметиазид, хлортиазид, гидрохлортиазид, гидрофлуметазид, метиклотиазид, политиазид, трихлорметиазид, хлорталидон, индапамид, метолазон, квинэтазон, ацетазоламид, дихлорфенамид, метазоламид, глицерин, изосорбид, маннитол, амилорид или триамтерен.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с антагонистом рецептора альдостерона или минералокортикоида, например, предпочтительно таким как спиронолактон или эплеренон.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с антагонистом рецептора вазопрессина, например, предпочтительно таким как кониваптан, толваптан, ликсиваптан или SR-121463.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с донором органического нитрата или NO, например, предпочтительно таким как нитропруссид натрия, нитроглицерин, изосорбидмононитрат, изосорбиддинитрат, молсидомин или SIN-1, или в комбинации с ингаляционным NO.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с соединением, обладающим позитивным инотропным действием, например, предпочтительно таким как сердечные гликозиды (дигоксин), а также бета-адренергические и дофаминергические агонисты, в частности изопротенеренол, адреналин, норадреналин, допамин или добутамин.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с антисимпатотоническими средствами, такими как резерпин, клонидин или альфа-метилдиоксифенилаланин, или в комбинации с агонистами калиевого канала, такими как миноксидил, диазоксид, дигидралазин или гидралазин.
Под средствами, которые обладают противосвертывающим действием, предпочтительно подразумевают соединения, выбранные из группы, включающей ингибиторы агрегирования тромбоцитов и антитромботические вещества.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с ингибитором агрегирования тромбоцитов, например, предпочтительно таким как аспирин, клопидогрел, тиклопидин или дипиридамол.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с ингибитором тромбина, например, предпочтительно таким как ксимелагатран, мелагатран, бивалирудин или клексан.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с антагонистом GPIIb/IIIa, например, предпочтительно таким как тирофибан или абциксимаб.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с ингибитором фактора Ха, например, предпочтительно таким как ривароксабан (BAY 59-7939), DU-176b, апиксабан, отамиксабан, фидексабан, разаксабан, фондапаринукс, идрапаринукс, PMD-3112, YM-150, KFA-1982, EMD-503982, МСМ-17, MLN-1021, DX 9065а, DPC 906, JTV 803, SSR-126512 или SSR-128428.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с гепарином или низкомолекулярным производным гепарина.
В предпочтительном варианте осуществления изобретения предлагаемые в изобретении соединения применяют в комбинации с антагонистами витамина К, например, предпочтительно такими как коумарин.
Под снижающими аритмию средствами предпочтительно подразумевают вещества из класса антиаритмических средств Ia (например, хинидин), класса антиаритмических средств Ic (например, флекаинид, пропафенон), класса антиаритмических средств II (например, метопролол, атенолол, соталол, окспренолол и другие ингибиторы бета-рецепторов), класса антиаритмических средств III (например, соталол, амиодарон) и класса антиаритмических средств IV (например, дигоксин, а также верапамил, дилтиазем и другие антагонисты кальция).
В соответствии с настоящим изобретением особенно предпочтительными являются комбинации, содержащие по меньшей мере одно из предлагаемых в изобретении соединений и одно или несколько других действующих веществ, выбранных из группы, включающей ингибиторы HMG-CoA-редуктазы (статин), диуретики, антагонисты бета-адреноцептора, антагонисты альфа-адреноцептора, доноры органических нитратов и NO, антагонисты кальция, ингибиторы АСЕ, антагонисты ангитензина АII, антагонисты рецептора альдостерона и минералокортикоида, антагонисты рецептора вазопрессина, ингибиторы агрегации тромбоцитов, противосвертывающие вещества и антиаритмические средства, а также их применение для лечения и/или профилактики указанных выше заболеваний.
Другим объектом настоящего изобретения являются лекарственные средства, содержащие по меньшей мере одно предлагаемое в изобретении соединение обычно совместно с одним или несколькими инертными, нетоксичными, фармацевтически пригодными вспомогательными веществами, а также их применение для указанных выше целей.
Предлагаемые в изобретении соединения могут обладать системным и/или местным действием. В соответствии с этим их можно применять, например, орально, парентерально, пульмонально, назально, сублингвально, лингвально, буккально, ректально, дермально, трансдермально, конъюнктивально, через слуховой проход или в виде имплантата, соответственно стента. Предлагаемые в изобретении соединения можно применять в формах, которые соответствуют указанным вариантам применения.
Для орального применения пригодны действующие в соответствии с уровнем техники формы применения, способные быстро и/или модифицированно высвобождать предлагаемые в изобретении соединения и содержащие предлагаемые в изобретении соединения в кристаллическом, аморфизованном и/или растворенном состоянии, например, такие как таблетки (таблетки без покрытия или снабженные, например, устойчивым к желудочному соку или медленно растворяющимся или нерастворимым покрытием, обеспечивающим контролируемое высвобождение предлагаемого в изобретении соединения), таблетки или пленки/лиофилизаты, быстро разрушающиеся в полости рта, пленки/облатки, капсулы (например, снабженные оболочкой из жесткого или мягкого желатина), драже, гранулы, пеллеты, порошки, эмульсии, суспензии, аэрозоли или растворы.
Парентеральное применение возможно в отсутствие резорбции (например, внутривенное, внутриартериальное, внутрисердечное, внутрипозвоночное или внутрипоясничное применение) или с резорбцией (например, внутримышечное, подкожное, внутрикожное, чрескожное или внутрибрюшинное применение). Пригодными для парентерального применения формами, в частности, являются препараты для инъекций и вливаний в виде растворов, суспензий, эмульсий, лиофилизатов или стерильных порошков.
Для прочих вариантов применения пригодны, например, ингаляционные лекарственные формы (в частности порошковые ингаляторы, аэрозольные распылители), нозальные капли, растворы или аэрозоли, таблетки для лингвального, сублингвального или буккального применения, пленки/облатки или капсулы, суппозитории, ушные или глазные препараты, вагинальные капсулы, водные суспензии (лосьоны, микстуры типа «болтушка»), липофильные суспензии, мази, кремы, трансдермальные терапевтические системы (например, пластыри), препараты в виде молочка, а также пасты, пены, присыпки, имплантаты или стенты.
Предпочтительным является оральное или парентеральное применение, прежде всего оральное применение.
Предлагаемые в изобретении соединения могут быть преобразованы в указанные выше формы применения. Соответствующие преобразования можно выполнять известными методами смешивания предлагаемых в изобретении соединений с инертными, нетоксичными, фармацевтически пригодными вспомогательными веществами. К пригодным вспомогательным веществам, в частности, относятся вещества-переносчики (например, микрокристаллическая целлюлоза, лактоза или маннитол), растворители (например, жидкие полиэтиленгликоли), эмульгаторы и диспергаторы или смачивающие агенты (например, додецилсульфат натрия или полиоксисорбитанолеат), связующие вещества (например, поливинилпирролидон), синтетические и природные полимеры (например, альбумин), стабилизаторы (в частности антиоксиданты, например, такие как аскорбиновая кислота), красители (в частности неорганические пигменты, например, такие как оксиды железа), а также вещества, предназначенные для корректирования вкуса и/или запаха.
В общем случае количество предлагаемого в изобретении соединения, необходимое для достижения желаемого эффекта, при парентеральном применении предпочтительно находится в примерном интервале от 0,001 до 1 мг, преимущественно от 0,01 до 0,5 мг на килограмм массы тела. При оральном применении соответствующая дозировка составляет примерно от 0,01 до 100 мг/кг, предпочтительно от 0,01 до 20 мг/кг и еще более предпочтительно от 0,1 до 10 мг/кг массы тела.
Несмотря на это, при необходимости, может потребоваться корректировка указанных выше дозировок, которая определяется массой тела, вариантом применения препарата, индивидуальным отношением пациента к действующему веществу, типом препарата и моментом времени его применения (периодичностью). Так, например, в некоторых случаях могут оказаться достаточными дозировки, более низкие по сравнению с указанными выше минимальными предельными значениями, тогда как в других случаях могут потребоваться дозировки, превышающие указанные выше максимальные значения. В последнем случае рекомендуется распределять дозировку на несколько применяемых в течение суток доз.
Приведенные ниже примеры служат для пояснения настоящего изобретения. Примеры не ограничивают объема изобретения.
В отсутствие особых указаний процентные данные в нижеследующих опытах и примерах приведены в массовых процентах, части в массовых частях. Количественные соотношения между компонентами растворителей, степени разбавления и концентрации растворов жидких компонентов в жидкостях указаны в соответствующих объемных единицах.
А. Примеры
Сокращения и аббревиатуры
|
Методы LC-MS и ВЭЖХ
Метод 1 (LC-MS). Тип масс-спектрометра: Micromass ZQ; тип прибора для ВЭЖХ: Waters Alliance 2795; колонка Phenomenex Synergi, 2 мкм Hydro-RP Mercury 20 мм × 4 мм; элюент А: 1 л воды + 0,5 мл 50-процентной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0,5 мл 50-процентной муравьиной кислоты; градиент: 0,0 мин 90% А→2,5 мин 30% А→3,0 мин 5% А→4,5 мин 5% А; расход: 0,0 мин 1 мл/мин, 2,5 мин/3,0 мин/4,5 мин 2 мл/мин; температура печи 50°С; УФ-детектирование 210 нм.
Метод 2 (LC-MS). Прибор: Micromass Quattro LCZ с ВЭЖХ Agilent Serie 1100; колонка: Phenomenex Synergi, 2 мкм Hydro-RP Mercury, 20 мм × 4 мм; элюент A: 1 л воды + 0,5 мл 50-процентной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0,5 мл 50-процентной муравьиной кислоты; градиент: 0,0 мин 90% А 2,5 мин 30% А→3,0 мин 5% А→4,5 мин 5% А; расход: 0,0 мин 1 мл/мин, 2,5 мин/3,0 мин/4,5 мин 2 мл/мин; температура печи 50°С; УФ-детектирование 208-400 нм.
Метод 3 (LC-MS). Тип масс-спектрометра: Micromass ZQ; тип прибора для ВЭЖХ: HP 1100 Series; УФ-детектор DAD; колонка Phenomenex Synergi, 2 мкм Hydro-RP Mercury, 20 мм × 4 мм; элюент А: 1 л воды + 0,5 мл 50-процентной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0,5 мл 50-процентной муравьиной кислоты; градиент: 0,0 мин 90% А→2,5 мин 30% А→3,0 мин 5% А→4,5 мин 5% А; расход: 0,0 мин 1 мл/мин, 2,5 мин/3,0 мин /4,5 мин 2 мл/мин; температура печи 50°С; УФ-детектирование 210 нм.
Метод 4 (LC-MS). Тип масс-спектрометра: Micromass ZQ; тип прибора для ВЭЖХ: HP 1100 Series; УФ-детектор DAD; колонка Phenomenex Gemini, 3 мкм, 30 мм × 3,00 мм; элюент А: 1 л воды + 0,5 мл 50-процентной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0,5 мл 50-процентной муравьиной кислоты; градиент: 0,0 мин 90% А→2,5 мин 30% А→3,0 мин 5% А→4,5 мин 5% А; расход: 0,0 мин 1 мл/мин, 2,5 мин/3,0 мин /4,5 мин 2 мл/мин; температура печи 50°С; УФ-детектирование 210 нм.
Метод 5 (препаративная ВЭЖХ). Тип прибора для ВЭЖХ: насос Abimed/Gilson 305/306; манометрический узел 806; УФ-узел: Knauer Variable Wavelength Monitor; колонка: Gromsil C18, 10 нм, 250 мм × 30 мм; элюент А: 1 л вода + 0,5 мл 99-процентной трифторуксусной кислоты, элюент В: 1 л ацетонитрила; градиент: 0,0 мин 2% В→10 мин 2% В→50 мин 90% В; расход 20 мл/мин; объем: 628 мл А и 372 мл В.
Метод 6а (препаративная ВЭЖХ). Колонка VP 250/21 Nukleodur 100-5 С18 ec, Macherey & Nagel Nr. 762002; элюент А: вода/0,01% трифторуксусной кислоты, элюент В: ацетонитрил/0,01% трифторуксусной кислоты; градиент: 0 мин 0% В→20 мин 20% В→40 мин 20% В→60 мин 30% В→80 мин 30% В→90 мин 100% В→132 мин 100% В; расход 5 мл/мин; комнатная температура; УФ-детектирование 210 нм.
Метод 6b (препаративная ВЭЖХ). Колонка VP 250/21 Nukleodur 100-5 С18 ес, Macherey & Nagel Nr. 762002; элюент A: 1 литр воды/1 мл 99-процентной трифторуксусной кислоты, элюент В: 1 литр ацетонитрила/1 мл 99-процентной трифторуксусной кислоты; градиент: 0 мин 30% В→20 мин 50% В→40 мин 80% В→60 мин 100% В; расход 5 мл/мин; комнатная температура; УФ-детектирование 210 нм.
Метод 7 (аналитическая ВЭЖХ). Колонка XTerra WAT 186000478, 3,9 мм × 150 мм; элюент А: 10 мл 70-процентной перхлорной кислоты в 2,5 л воды, элюент В: ацетонитрил; градиент: 0,0 мин 20% В→1 мин 20% В→4 мин 90% В→9 мин 90% В; комнатная температура; расход 1 мл/мин.
Метод 8 (LC-MS). Масс-спектрометр Micromass Quattro LCZ с ВЭЖХ Agilent серии 1100; колонка Phenomenex Onyx Monolithic С18, 100 мм × 3 мм; элюент А: 1 л воды + 0,5 мл 50-процентной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0,5 мл 50-процентной муравьиной кислоты; градиент: 0,0 мин 90% А→2 мин 65% А→4,5 мин 5% А→6 мин 5% А; расход: 2 мл/мин; температура печи 40°С; УФ-детектирование 208-400 нм.
Метод 9 (LC-MS). Прибор: Micromass Platform LCZ с ВЭЖХ Agilent Serie 1100; колонка Thermo Hypersil GOLD, 3 мкм, 20 мм × 4 мм; элюент А: 1 л воды + 0,5 мл 50-процентной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0,5 мл 50-процентной муравьиной кислоты; градиент: 0,0 мин 100% А→0,2 мин 100% А→2,9 мин 30% А→3,1 мин 10% А→5,5 мин 10% А; температура печи 50°С; расход: 0,8 мл/мин; УФ-детектирование 210 нм.
Метод 10 (LC-MS). Тип масс-спектрометра: Micromass ZQ; тип прибора ВЭЖХ: HP серии 1100; УФ-детектор DAD; колонка Phenomenex Gemini, 3 мкм, 30 мм × 3,00 мм; элюент А: 1 л воды + 0,5 мл 50-процентной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0,5 мл 50-процентной муравьиной кислоты; градиент: 0,0 мин 90% А→2,5 мин 30% А→3,0 мин 5% А→4,5 мин 5% А; расход 0,0 мин 1 мл/мин, 2,5 мин/3,0 мин/4,5 мин 2 мл/мин; температура печи 50°С; УФ-детектирование 210 нм.
Метод 11 (LC-MS). Тип масс-спектрометра: Micromass ZQ; тип прибора ВЭЖХ: Waters Alliance 2795; колонка Phenomenex Synergi, 2,5 мкм MAX-RP 100А Mercury, 20 мм × 4 мм; элюент А: 1 л воды + 0,5 мл 50-процентной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0,5 мл 50-процентной муравьиной кислоты; градиент: 0,0 мин 90% А→0,1 мин 90% А→3,0 мин 5% А→4,0 мин 5% А→4,01 мин 90% А; расход 2 мл/мин; температура печи 50°С; УФ-детектирование 210 нм.
Метод 12 (LC-MS). Прибор: Micromass Quattro LCZ с ВЭЖХ Agilent Serie 1100; колонка: Phenomenex Synergi, 2,5 мкм MAX-RP 100A Mercury, 20 мм × 4 мм; элюент A: 1 л воды + 0,5 мл 50-процентной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0,5 мл 50-процентной муравьиной кислоты; градиент: 0,0 мин 90% А→0,1 мин 90% А→3,0 мин 5% А→4,0 мин 5% А→4,1 мин 90% А; температура печи 50°С; УФ-детектирование 208-400 нм.
Метод 13 (LC-MS). Тип масс-спектрометра: Micromass Quattro Premier с Waters UPLC Acquity; колонка: Thermo Hypersil GOLD, 1,9 мкм, 50 мм × 1 мм; элюент A: 1 л воды + 0,5 мл 50-процентной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0,5 мл 50-процентной муравьиной кислоты; градиент: 0,0 мин 90% А→0,1 мин 90% А→1,5 мин 10% А→2,2 мин 10% А; температура печи 50°С; расход 0,33 мл/мин; УФ-детектирование 210 нм.
Исходные соединения и промежуточные продукты
Пример 1А
2-{4-[2-Амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}тио)-3,5-дицианопиридин-4-ил]фенокси}этил-L-валинат трифторацетат
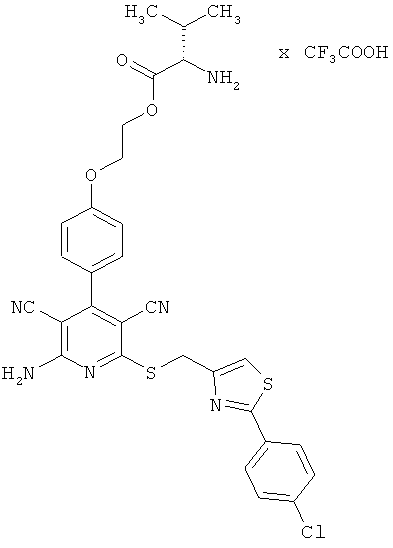
1 г (1,92 ммоль) 2-амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}тио)-4-[4-(2-гидроксиэтокси)фенил]пиридин-3,5-дикарбонитрила [согласно примеру 6 из международной заявки WO 03/053441], 0,460 г (2,11 ммоль) N-Boc-L-валина, 0,442 г (2,31 ммоль) 1-(3-диметиламинопропил)-3-этилкар-бодиимид гидрохлорида, а также 0,023 г (0,19 ммоль) 4-диметиламинопиридина совмещают в смеси 40 мл дихлорметана с 10 мл ДМФ и в течение ночи перемешивают при комнатной температуре. Образуется прозрачный раствор. Продукты реакции выливают в смесь полунасыщенного раствора хлорида аммония с дихлорметаном. Органическую фазу отделяют, последовательно промывают водой, насыщенным раствором гидрокарбоната натрия и насыщенным раствором хлорида натрия, сушат над сульфатом магния, фильтруют и концентрируют. Остаток подвергают очистке флэш-хроматографией на силикагеле, используя в качестве элюента смесь дихлорметана с этилацетатом (градиент 10:1→7:1→5:1). Соответствующие фракции объединяют, растворитель удаляют отгонкой в вакууме. В результате сушки остатка в высоком вакууме получают 0,85 г Вос-защищенного промежуточного продукта (выход от теоретического 62%).
Остаток вводят в смесь 5 мл дихлорметана с 5 мл безводной трифторуксусной кислоты, и полученный раствор в течение двух часов перемешивают при комнатной температуре. Затем его концентрируют до сухого остатка, который смешивают с этилацетатом. Образующийся осадок выделяют фильтрованием, промывают диэтиловым эфиром и сушат в высоком вакууме. Получают 935 мг целевого соединения в виде бесцветных кристаллов (количественный выход).
ВЭЖХ (метод 7): Rt=5,5 мин.
LC-MS (метод 10): Rt=2,06 мин; MS (ESIpos): m/z=619 (М+Н)+.
Пример 2А
2-{4-[2-Амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}тио)-3,5-дицианопиридин-4-ил]фенокси}этил-L-аланинат трифторацетат
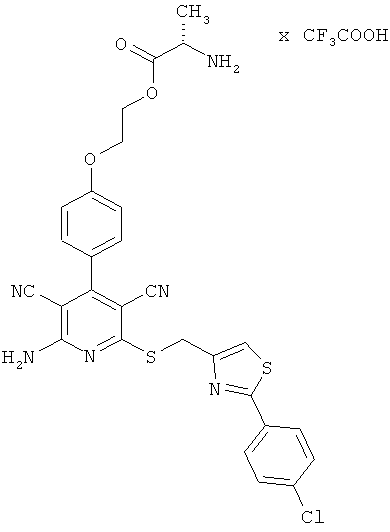
1,5 г (2,88 ммоль) 2-амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}-тио)-4-[4-(2-гидроксиэтокси)фенил]пиридин-3,5-дикарбонитрила, 1,64 г (8,66 ммоль) N-Boc-L-аланина, 0,719 г (3,75 ммоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида, а также 0,176 г (1,44 ммоль) 4-диметиламинопиридина совмещают в смеси 25 мл дихлорметана с 25 мл ДМФ и в течение двух часов перемешивают при комнатной температуре. Реакционные продукты концентрируют в вакууме, остаток вводят в этилацетат. Смесь экстрагируют дважды 5-процентной лимонной кислотой и дважды раствором гидрокарбоната натрия. Органическую фазу концентрируют, остаток смешивают с 50 мл диэтилового эфира, а также с 50 мл пентана. Остаток отсасывают на вакуум-фильтре и промывают пентаном. В результате сушки остатка в высоком вакууме получают 1,23 г Вос-защищенного промежуточного продукта (выход от теоретического 62%).
Остаток вводят в смесь 18 мл дихлорметана с 2 мл безводной трифторуксусной кислоты, и раствор в течение часа обрабатывают при комнатной температуре в ультразвуковой ванне. Затем его концентрируют, и остаток смешивают с диэтиловым эфиром. Образующийся осадок выделяют фильтрованием, промывают диэтиловым эфиром и сушат в высоком вакууме. Получают 1200 мг целевого соединения в виде бесцветных кристаллов (выход от теоретического 96%).
ВЭЖХ (метод 7): Rt=5,3 мин.
LC-MS (метод 12): Rt=1,73 мин; MS (ESIpos): m/z=591 (М+Н)+.
Пример 3А
2-{4-[2-Амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}тио)-3,5-дицианопиридин-4-ил]фенокси}этил-O-трет-бутил-L-серинат
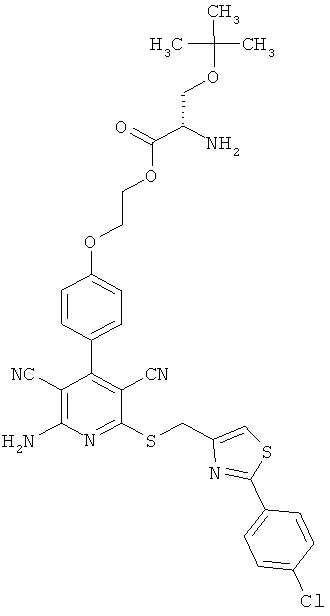
1 г (1,92 ммоль) 2-амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}тио)-4-[4-(2-гидроксиэтокси)фенил]пиридин-3,5-дикарбонитрила, 0,612 г (2,12 ммоль) N-(трет-бутоксикарбонил)-O-трет-бутил-L-серина, 0,442 г (2,31 ммоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида, а также 0,024 г (0,192 ммоль) 4-диметиламинопиридина совмещают в смеси 40 мл дихлорметана с 10 мл ДМФ и в течение ночи перемешивают при комнатной температуре. Затем реакционные продукты выливают в смесь полунасыщенного раствора хлорида аммония с дихлорметаном. Органическую фазу отделяют, последовательно промывают водой, насыщенным раствором гидрокарбоната натрия и насыщенным раствором хлорида натрия, сушат над сульфатом магния, фильтруют и концентрируют. Остаток вводят в дихлорметан и смешивают с диэтиловым эфиром. Образующийся осадок отсасывают на вакуум-фильтре и промывают диэтиловым эфиром. В результате сушки осадка в высоком вакууме получают 1,25 г защищенного промежуточного продукта (выход от теоретического 85%).
Остаток вводят в смесь 100 мл дихлорметана с 10 мл безводной трифторуксусной кислоты, и раствор в течение часа перемешивают при комнатной температуре. Затем реакционные продукты выливают в смесь полунасыщенного раствора гидрокарбоната натрия с дихлорметаном. Органическую фазу отделяют, сушат над сульфатом магния, фильтруют и концентрируют. В результате сушки остатка в высоком вакууме получают 1020 мг целевого соединения в виде бесцветного порошка (выход от теоретического 95%).
ВЭЖХ (метод 7): Rt=5,4 мин.
LC-MS (метод 11): Rt=1,65 мин; MS (ESIpos): m/z=663 (М+Н)+.
Пример 4А
N2-(трет-бутоксикарбонил)-N-{2-[(трет-бутоксикарбонил)амино]этил}-L-α-глутамин
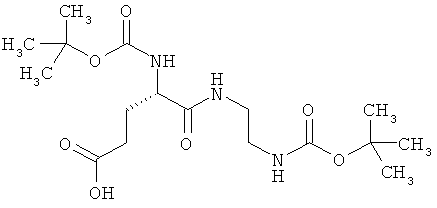
1,5 г (4,45 ммоль) (2S)-5-(бензилокси)-2-[(трет-бутоксикарбонил)амино]-5-оксопентановой кислоты, 783 мг (4,89 ммоль) трет-бутил-(2-аминоэтил)-карбамата, 938 мг (4,89 ммоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида, а также 749 мг (0,489 ммоль) 1-гидрокси-1Н-бензотриазол гидрата совмещают в 140 мл ДМФ и в течение ночи перемешивают при комнатной температуре. Затем реакционные продукты выливают в смесь полунасыщенного раствора хлорида аммония с этилацетатом. Органическую фазу отделяют, последовательно промывают водой, насыщенным раствором гидрокарбоната натрия и насыщенным раствором хлорида натрия, сушат над сульфатом магния, фильтруют и концентрируют. Остаток смешивают с диэтиловым эфиром. Образующийся осадок отсасывают на вакуум-фильтре, промывают диэтиловым эфиром и сушат в высоком вакууме. Получают 1,9 г защищенного промежуточного продукта (выход от теоретического 89%).
LC-MS (метод 12): Rt=2,19 мин;
MS (ESIpos): m/z=480 (М+Н)+.
1,9 г (3,96 ммоль) полученного промежуточного продукта растворяют в 125 мл метанола и после добавления 250 мг 10-процентного палладия на активированном угле в течение двух часов гидрируют при комнатной температуре и нормальном давлении. Затем катализатор отделяют фильтрованием, и растворитель удаляют отгонкой в вакууме. Получают 1500 мг целевого соединения в виде бесцветной пены (выход от теоретического 97%).
LC-MS (метод 10): Rt=1,94 мин; MS (ESIpos): m/z=390 (М+Н)+.
Пример 5А
N2-(трет-бутоксикарбонил)-N-{2-[(трет-бутоксикарбонил)амино]этил}-L-глутамин
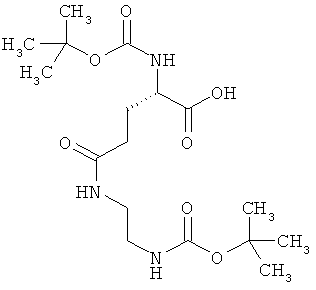
1,5 г (4,45 ммоль) (4S)-5-(бензилокси)-4-[(трет-бутоксикарбонил)амино]-5-оксопентановой кислоты, 783 мг (4,89 ммоль) трет-бутил-(2-аминоэтил)-карбамата, 938 мг (4,89 ммоль) 1-(3-диметиламинопропил)-3-этилкарбо-диимид гидрохлорида, а также 749 мг (4,89 ммоль) 1-гидрокси-1Н-бензотриазол гидрата совмещают в 140 мл ДМФ и в течение ночи перемешивают при комнатной температуре. Затем реакционные продукты выливают в смесь полунасыщенного раствора хлорида аммония с этилацетатом. Органическую фазу отделяют, последовательно промывают водой, насыщенным раствором гидрокарбоната натрия и насыщенным раствором хлорида натрия, сушат над сульфатом магния, фильтруют и концентрируют. Остаток смешивают с диэтиловым эфиром. Образующийся осадок отсасывают на вакуум-фильтре, промывают диэтиловым эфиром и сушат в высоком вакууме. Получают 2,1 г защищенного промежуточного продукта (выход от теоретического 98%).
LC-MS (метод 10): Rt=2,47 мин; MS (ESIpos): m/z=480 (М+Н)+.
2,1 г (4,38 ммоль) полученного промежуточного продукта растворяют в 140 мл метанола и после добавления 250 мг 10-процентного палладия на активированном угле в течение двух часов гидрируют при комнатной температуре и нормальном давлении. Затем катализатор отделяют фильтрованием, и растворитель удаляют отгонкой в вакууме. Получают 1540 мг целевого соединения в виде бесцветной пены (выход от теоретического 90%).
LC-MS (метод 11): Rt=1,35 мин; MS (ESIpos): m/z=390 (М+Н)+.
Пример 6А
N2-(трет-бутоксикарбонил)-N-{2-[(трет-бутоксикарбонил)амино]этил}-L-аспарагин
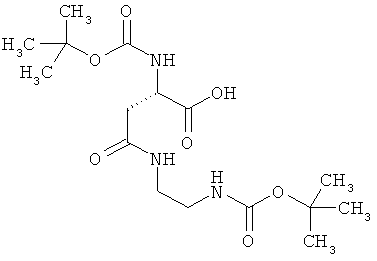
1,5 г (4,64 ммоль) (3S)-4-(бензилокси)-3-[(трет-бутоксикарбонил)амино]-4-оксомасляной кислоты, 818 мг (5,1 ммоль) трет-бутил-(2-аминоэтил)-карбамата, 978 мг (5,1 ммоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида, а также 781 мг (5,1 ммоль) 1-гидрокси-1Н-бензотриазол гидрата совмещают в 75 мл ДМФ и в течение ночи перемешивают при комнатной температуре. Затем реакционные продукты выливают в смесь полунасыщенного раствора хлорида аммония с этилацетатом. Органическую фазу отделяют, последовательно промывают водой, насыщенным раствором гидрокарбоната натрия и насыщенным раствором хлорида натрия, сушат над сульфатом магния, фильтруют и концентрируют. Остаток смешивают с диэтиловым эфиром. Образующийся осадок отсасывают на вакуум-фильтре, промывают диэтиловым эфиром и сушат в высоком вакууме. Получают 2,1 г защищенного промежуточного продукта (выход от теоретического 81%).
LC-MS (метод 10): Rt=2,47 мин; MS (ESIpos): m/z=466 (М+Н)+.
2,1 г (4,51 ммоль) полученного промежуточного продукта растворяют в 140 мл метанола и после добавления 250 мг 10-процентного палладия на активированном угле в течение двух часов гидрируют при комнатной температуре и нормальном давлении. Затем катализатор отделяют фильтрованием, и растворитель удаляют отгонкой в вакууме. Получают 1690 мг целевого соединения в виде бесцветной пены (выход от теоретического 99%).
LC-MS (метод 11): Rt=1,35 мин; MS (ESIpos): m/z=376 (М+Н)+.
Пример 7А
5-(2-{4-[2-Амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}тио)-3,5-дицианопиридин-4-ил]фенокси}этил)-1-трет-бутил-L-глутамат
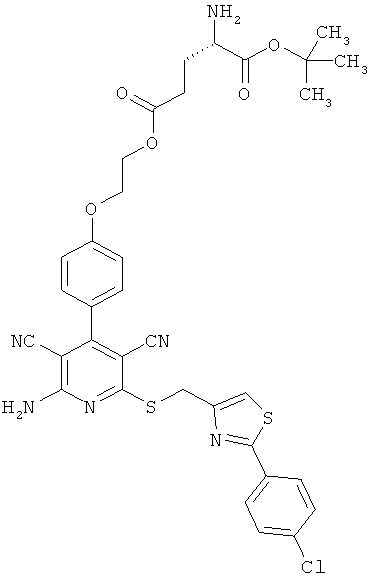
3,117 г (6 ммоль) 2-амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}тио)-4-[4-(2-гидроксиэтокси)фенил]пиридин-3,5-дикарбонитрила, 2 г (6,59 ммоль) (4S)-5-трет-бутокси-4-[(трет-бутоксикарбонил)амино]-5-оксопентаноата, 1,38 г (7,19 ммоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида, а также 0,073 г (0,6 ммоль) 4-диметиламинопиридина совмещают в смеси 80 мл дихлорметана с 20 мл ДМФ и в течение ночи перемешивают при комнатной температуре. Затем реакционные продукты выливают в смесь полунасыщенного раствора хлорида аммония с дихлорметаном. Органическую фазу отделяют, последовательно промывают водой, насыщенным раствором гидрокарбоната натрия и насыщенным раствором хлорида натрия, сушат над сульфатом магния, фильтруют и концентрируют. Остаток выделяют осаждением из смеси дихлорметана с петролейным эфиром. Продукт отсасывают на вакуум-фильтре, промывают диэтиловым эфиром и сушат в высоком вакууме. Получают 4,44 г защищенного промежуточного продукта (выход от теоретического 92%).
LC-MS (метод 10): Rt=3,38 мин; MS (ESIpos): m/z=805 (М+Н)+.
57 мг (0,07 ммоль) полученного промежуточного продукта вводят в смесь 6 мл дихлорметана с 0,6 мл безводной трифторуксусной кислоты, и раствор в течение 2,5 часов перемешивают при комнатной температуре. Затем реакционные продукты выливают в смесь полунасыщенного раствора гидрокарбоната натрия с дихлорметаном. Органическую фазу отделяют, сушат над сульфатом магния, фильтруют и концентрируют. В результате сушки в высоком вакууме получают 50 мг целевого соединения в виде бесцветного порошка (количественный выход).
ВЭЖХ (метод 7): Rt=5,4 мин.
LC-MS (метод 11): Rt=1,72 мин; MS (ESIpos): m/z=705 (М+Н)+.
Пример 8А
2-{4-[2-Амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}сульфанил)-3,5-дицианопиридин-4-ил]фенокси}этил-L-лейцинат трифторацетат
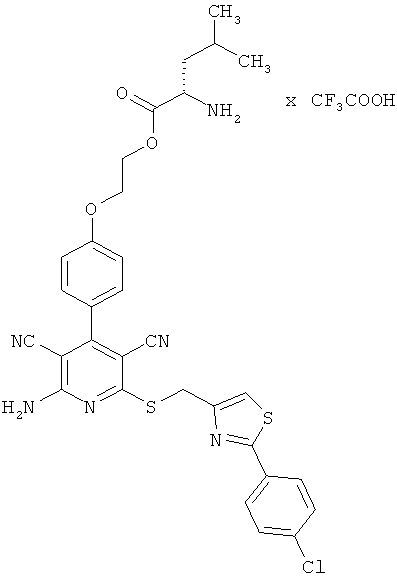
Целевое соединение получают аналогично примеру 1А из 2-амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}тио)-4-[4-(2-гидроксиэтокси)фенил]-пиридин-3,5-дикарбонитрила и Boc-L-лейцина.
ВЭЖХ (метод 7): Rt=5,5 мин.
LC-MS (метод 12): Rt=1,75 мин; MS (ESIpos): m/z=633 (M+H)+.
Пример 9А
2-{4-[2-Амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}сульфанил)-3,5-дицианопиридин-4-ил]фенокси}этил-D-аланинат трифторацетат
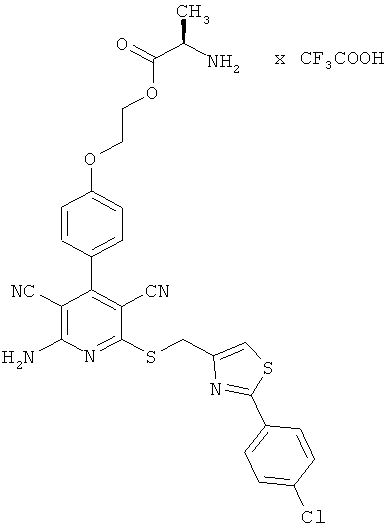
Целевое соединение получают аналогично примеру 2А из 2-амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}тио)-4-[4-(2-гидроксиэтокси)фенил]-пиридин-3,5-дикарбонитрила и Boc-D-аланина.
ВЭЖХ (метод 7): Rt=5,2 мин.
LC-MS (метод 13): Rt=1,15 мин; MS (ESIpos): m/z=591 (М+Н)+.
Пример 10А
N2-(трет-бутоксикарбонил)-N-{2-[(трет-бутоксикарбонил)амино]этил}-D-α-глутамин
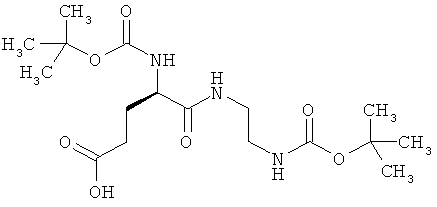
Целевое соединение получают аналогично примеру 4А из (2R)-5-(бензилокси)-2-[(трет-бутоксикарбонил)амино]-5-оксопентановой кислоты.
ВЭЖХ (метод 7): Rt=4,4 мин.
LC-MS (метод 11): Rt=1,37 мин; MS (ESIpos): m/z=390 (М+Н)+.
Пример 11А
2-{4-[2-Амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}сульфанил)-3,5-дицианопиридин-4-ил]фенокси}этилглицинат трифторацетат
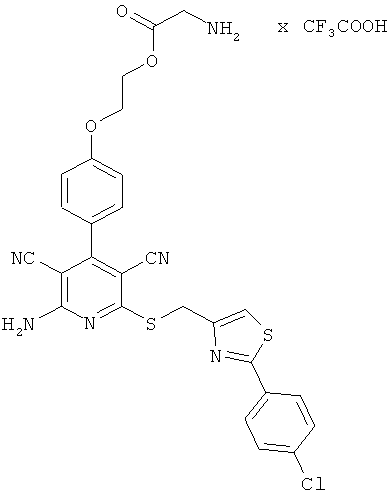
Целевое соединение получают аналогично примеру 2А из 2-амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}тио)-4-[4-(2-гидроксиэтокси)фенил]-пиридин-3,5-дикарбонитрила и Вос-глицина.
ВЭЖХ (метод 7): Rt=5,1 мин.
LC-MS (метод 13): Rt=1,13 мин; MS (ESIpos): m/z=577 (М+Н)+.
Пример 12А
2-{4-[2-Амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}сульфанил)-3,5-дицианопиридин-4-ил]фенокси}этил-L-фенилаланинат трифторацетат
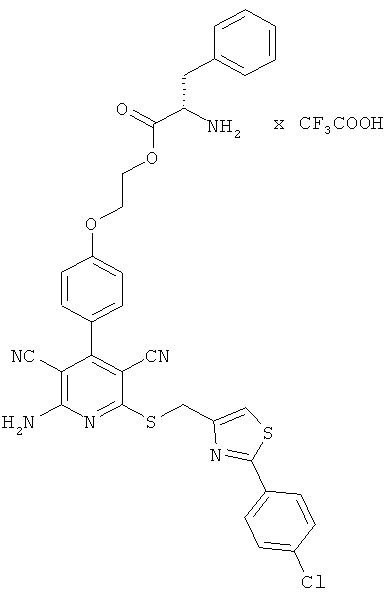
Целевое соединение получают аналогично примеру 2А из 2-амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}тио)-4-[4-(2-гидроксиэтокси)фенил]-пиридин-3,5-дикарбонитрила и Boc-L-фенилаланина.
ВЭЖХ (метод 7): Rt=5,1 мин.
LC-MS (метод 13): Rt=1,13 мин; MS (ESIpos): m/z=577 (М+Н)+.
Примеры осуществления изобретения
Пример 1
2-{4-[2-Амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}тио)-3,5-дицианопиридин-4-ил]фенокси}этил-L-лизил-L-валинат дигидрохлорид
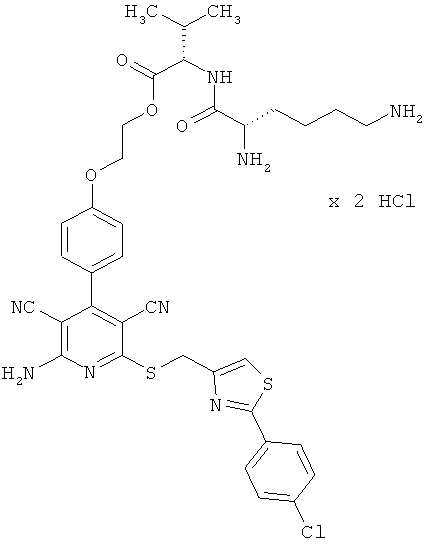
1,5 г (1,77 ммоль) соединения из примера 1А, 2,36 г (5,31 ммоль) 2,5-диоксопирролидин-1-ил-N2,N6-бис(трет-бутоксикарбонил)-L-лизината и 1,5 мл N,N-диизопропилэтиламина совмещают в 20 мл ДМФ и в течение ночи перемешивают при комнатной температуре. Затем растворитель удаляют отгонкой в вакууме, и остаток подвергают очистке флэш-хроматографией на силикагеле, используя в качестве элюента смесь дихлорметана с этилацетатом (градиент 3:1→2:1). Соответствующие фракции объединяют, и растворитель удаляют отгонкой в вакууме. В результате сушки остатка в высоком вакууме получают 1,2 г защищенного промежуточного продукта (выход от теоретического 66%).
ВЭЖХ (метод 7): Rt=6,7 мин.
1,2 г (1,27 ммоль) полученного промежуточного продукта вводят в 3 мл дихлорметана и смешивают с 50 мл насыщенного раствора водородхлорида в дихлорметане. Реагенты в течение 30 минут перемешивают при комнатной температуре, при этом целевой продукт выпадает в осадок. Растворитель удаляют выпариванием, остаток смешивают с 70 мл диэтилового эфира. Смесь фильтруют, остаток на фильтре промывают диэтиловым эфиром и сушат в высоком вакууме. Получают 893 мг целевого соединения в виде бесцветных кристаллов (выход от теоретического 86%).
ВЭЖХ (метод 7): Rt=5,1 мин.
LC-MS (метод 12): Rt=1,47 мин; MS (ESIpos): m/z=747 (М+Н)+.
1Н-ЯМР (400 МГц, ДМСО-d6): δ=0,94 и 0,95 (2d, 6Н), 1,4 (m, 2Н), 1,55 (m, 2Н), 1,75 (m, 2Н), 2,14 (m, 1Н), 2,7-2,8 (m, 2Н), 3,95 (m, 1Н), 4,3-4,5 (m, 2Н), 4,65 (s, 2Н), 7,12 (d, 2Н), 7,51 (d, 2Н), 7,58 (d, 2Н), 7,95 (d, 2Н), 7,97 (s, 1Н), 8,0 (m, 2Н), 8,3 (m, 2Н), 8,8 (d, 1Н).
Пример 2
2-{4-[2-Амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}тио)-3,5-дицианопиридин-4-ил]фенокси}этил-β-аланил-L-валинат гидрохлорид
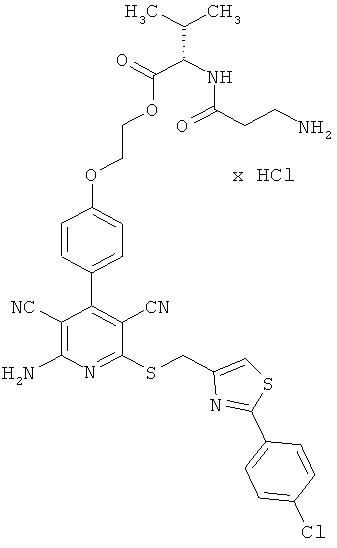
0,1 г (0,12 ммоль) соединения из примера 1А, 0,051 г (0,18 ммоль) 2,5-диоксопирролидин-1-ил-N-(трет-бутоксикарбонил)-β-аланината и 82 мкл N,N-диизопропилэтиламина совмещают в 6 мл ДМФ и в течение ночи перемешивают при комнатной температуре. Затем растворитель удаляют отгонкой в вакууме, и остаток подвергают очистке флэш-хроматографией на силикагеле, используя в качестве элюента смесь дихлорметана с этилацетатом (градиент 4:1→3:1→2:1). Соответствующие фракции объединяют, и растворитель удаляют отгонкой в вакууме. В результате сушки остатка в высоком вакууме получают 0,052 г защищенного промежуточного продукта (выход от теоретического 56%).
ВЭЖХ (метод 7): Rt=6,3 мин.
0,05 г (0,063 ммоль) полученного промежуточного продукта вводят в 1 мл дихлорметана и смешивают с 15 мл насыщенного раствора водородхлорида в дихлорметане. Реагенты перемешивают в течение трех часов при комнатной температуре, при этом целевое соединение выпадает в осадок. Растворитель удаляют выпариванием, остаток смешивают с 10 мл диэтилового эфира. Смесь фильтруют, остаток на фильтре промывают диэтиловым эфиром и сушат в высоком вакууме. Получают 41 мг целевого соединения в виде бесцветных кристаллов (выход от теоретического 85%).
ВЭЖХ (метод 7): Rt=5,2 мин.
LC-MS (метод 12): Rt=2,1 мин; MS (ESIpos): m/z=690 (М+Н)+.
1Н-ЯМР (400 МГц, ДМСО-d6): δ=0,89 и 0,9 (2d, 6Н), 2,04 (m, 2Н), 2,5 (m, 2Н), 2,9-3,0 (m, 2Н), 4,2 (m, 1Н), 4,25 (m, 2Н), 4,35-4,5 (m, 2Н), 4,67 (s, 2Н), 7,12 (d, 2Н), 7,5 (d, 2Н), 7,57 (d, 2Н), 7,8 (m, 2Н), 7,94 (s, 1Н), 7,95 (d, 2Н), 8,5 (d, 1Н).
Пример 3
2-{4-[2-Амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}тио)-3,5-дицианопиридин-4-ил]фенокси}этил-L-аргинил-L-валинат дигидрохлорид
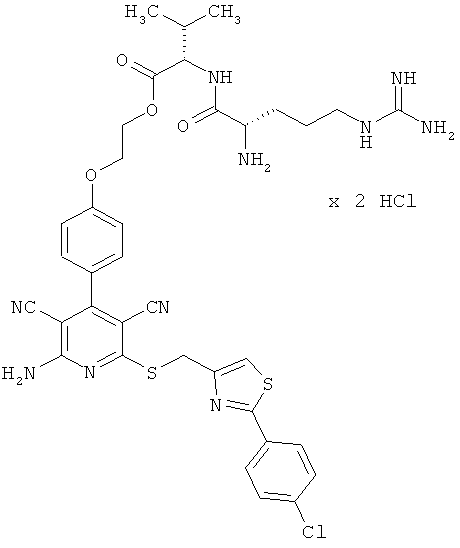
2,24 г (4,72 ммоль) N2,N5-бис(трет-бутоксикарбонил)-N5-{[(трет-бутокси-карбонил)амино](имино)метил}-L-орнитина, 0,96 г (7,08 ммоль) 1-гидрокси-1Н-бензотриазол гидрата и 1,09 г (5,66 ммоль) N-(3-диметиламинопропил)-N'-этилкарбодиимид гидрохлорида совмещают в 200 мл ДМФ. После 30-минутного перемешивания добавляют 2 г (2,36 ммоль) соединения из примера 1А, а также 1,65 мл N,N-диизопропилэтиламина, и компоненты в течение ночи перемешивают при комнатной температуре. Затем реакционную смесь концентрируют, остаток смешивают с водой. Смесь отсасывают на вакуум-фильтре, и остаток подвергают очистке флэш-хроматографией на силикагеле, используя в качестве элюента смесь дихлорметана с этилацетатом (градиент 4:1→3:1). Соответствующие фракции объединяют, и растворитель удаляют отгонкой в вакууме. В результате сушки остатка в высоком вакууме получают 1,12 г защищенного промежуточного продукта (выход от теоретического 44%).
ВЭЖХ (метод 7): Rt=6,1 мин.
1,12 г (1,04 ммоль) промежуточного продукта вводят в 10 мл дихлорметана, смешивают с 10 мл безводной трифторуксусной кислоты и в течение ночи перемешивают при комнатной температуре. Затем реакционные продукты концентрируют, и остаток дважды промывают ТГФ. Смесь фильтруют, остаток на фильтре вводят в смесь 25 мл ТГФ с 5 мл метанола. При перемешивании добавляют 20 мл 2М раствора водородхлорида в диэтиловом эфире. Образующийся после кратковременного дополнительного перемешивания осадок отсасывают на вакуум-фильтре и промывают диэтиловым эфиром. В результате сушки в высоком вакууме получают 920 мг целевого соединения в виде бесцветных кристаллов (выход от теоретического 99%).
ВЭЖХ (метод 7): Rt=5,1 мин.
LC-MS (метод 10): Rt=1,7 мин; MS (ESIpos): m/z=775 (М+Н)+.
1Н-ЯМР (400 МГц, ДМСО-d6): δ=0,95 и 0,97 (2d, 6Н), 1,6 (m, 2Н), 1,75 (m, 2Н), 2,14 (m, 1Н), 3,25 (m, 2Н), 4,05 (m, 1Н), 4,25 (t, 1Н), 4,3 (m, 2Н), 4,4-4,5 (2m, 2Н), 4,65 (s, 2Н), 7,12 (d, 2Н), 7,48 (d, 2Н), 7,58 (d, 2Н), 7,95 (m, 3H), 8,4 (m, 2Н), 8,9 (d, 1Н).
Пример 4
2-{4-[2-Амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}тио)-3,5-дицианопиридин-4-ил]фенокси}этил-L-лизил-L-аланинат дигидрохлорид
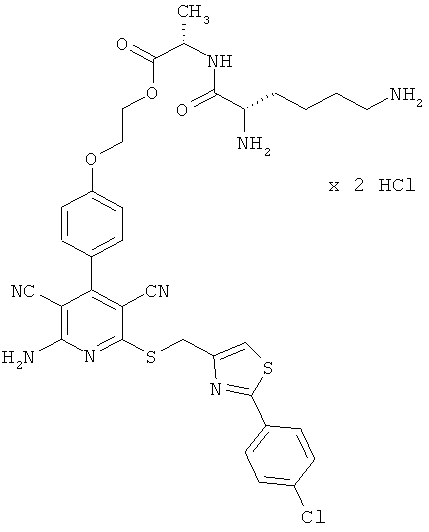
1,2 г (1,7 ммоль) соединения из примера 2А, 1,13 г (2,55 ммоль) 2,5-диоксопирролидин-1-ил-N2,N6-бис(трет-бутоксикарбонил)-L-лизината и 1,5 мл N,N-диизопропилэтиламина совмещают в 40 мл ДМФ и в течение ночи перемешивают при комнатной температуре. Затем продукты реакции концентрируют, и остаток распределяют между этилацетатом и водой. Органическую фазу отделяют и последовательно экстрагируют дважды 5-процентной лимонной кислотой и дважды 5-процентным раствором гидрокарбоната натрия. Органическую фазу концентрируют, и остаток подвергают очистке флэш-хроматографией на силикагеле, используя в качестве элюента смесь дихлорметана с этилацетатом (градиент 3:1 → 2:1). Соответствующие фракции объединяют, и растворитель удаляют отгонкой в вакууме. Остаток смешивают с 50 мл диэтилового эфира, а также с 50 мл пентана, и отсасывают на вакуум-фильтре. В результате сушки остатка в высоком вакууме получают 1,14 г защищенного промежуточного продукта (выход от теоретического 73%).
ВЭЖХ (метод 7): Rt=6,4 мин.
LC-MS (метод 11): Rt=2,64 мин; MS (ESIpos): m/z=919 (М+Н)+.
1,14 г (1,24 ммоль) промежуточного продукта вводят в 10 мл дихлорметана и смешивают с 60 мл насыщенного раствора водородхлорида в дихлорметане. Продукты реакции в течение ночи перемешивают при комнатной температуре, при этом целевое соединение выпадает в осадок. Растворитель концентрируют до одной трети от первоначального объема, и образовавшуюся суспензию смешивают с 200 мл диэтилового эфира. Смесь фильтруют, остаток на фильтре промывают диэтиловым эфиром и сушат в высоком вакууме. Получают 1,0 г целевого соединения в виде бесцветных кристаллов (количественный выход).
ВЭЖХ (метод 7): Rt=5,0 мин.
LC-MS (метод 10): Rt=1,67 мин; MS (ESIpos): m/z=719 (М+Н)+.
1Н-ЯМР (400 МГц, ДМСО-d6): δ=1,35 (d, 3H), 1,4 (m, 2Н), 1,6 (m, 2Н), 1,75 (m, 2Н), 2,75 (m, 2Н), 3,8 (m, 1Н), 4,25 (m, 2Н), 4,3-4,5 (m, 3H), 4,63 (s, 2Н), 7,12 (d, 2Н), 7,48 (d, 2Н), 7,58 (d, 2Н), 7,9-8,0 (m, 5Н), 8,3 (m, 2Н), 9,05 (d, 1Н).
Пример 5
2-{4-[2-Амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}тио)-3,5-дицианопиридин-4-ил]фенокси}этил-L-лизил-L-серинат дигидрохлорид
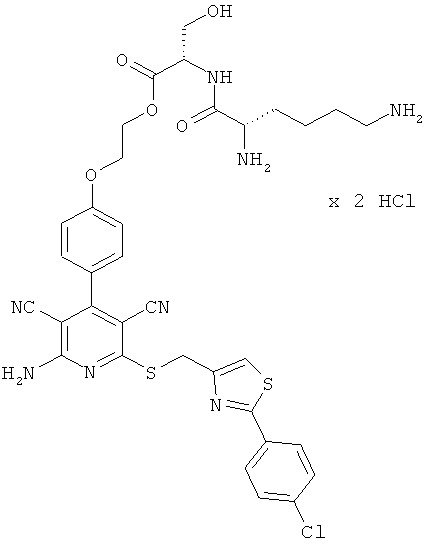
0,58 г (1,675 ммоль) N2,N6-бис(трет-бутоксикарбонил)-L-лизина, 0,28 г (1,83 ммоль) 1-гидрокси-1Н-бензотриазол гидрата и 0,35 г (1,83 ммоль) N-(3-диметиламинопропил)-N'-этилкарбодиимид гидрохлорида совмещают в 40 мл ДМФ и смешивают с 1,01 г (1,52 ммоль) соединения из примера 3А. Реагенты в течение ночи перемешивают при комнатной температуре и выливают в смесь полунасыщенного раствора хлорида аммония с дихлорметаном. Органическую фазу отделяют, последовательно промывают водой, насыщенным раствором гидрокарбоната натрия и насыщенным раствором хлорида натрия, сушат над сульфатом магния, фильтруют и концентрируют. Остаток подвергают очистке флэш-хроматографией на силикагеле, используя в качестве элюента сначала смесь дихлорметана с этилацетатом (градиент 3:1→2:1), а затем смесь дихлорметан/этилацетат/метанол (150:50:5). Соответствующие фракции объединяют и концентрируют. В результате сушки остатка в высоком вакууме получают 1,23 г защищенного промежуточного продукта (выход от теоретического 81%).
ВЭЖХ (метод 7): Rt=6,5 мин.
LC-MS (метод 12): Rt=2,99 мин; MS (ESIpos): m/z=991 (М+Н)+.
1,216 г (1,48 ммоль) промежуточного продукта вводят в 6 мл дихлорметана, смешивают с 6 мл безводной трифторуксусной кислоты, и реагенты в течение ночи перемешивают при комнатной температуре. Реакционные продукты концентрируют, остаток вновь смешивают с дихлорметаном. Смесь фильтруют, и остаток на фильтре вводят в смесь 25 мл дихлорметана с 25 мл этилацетата. При перемешивании добавляют 20 мл 2М раствора водородхлорида в диэтиловом эфире. Образующийся после кратковременного дополнительного перемешивания осадок отсасывают на вакуум-фильтре, промывают диэтиловым эфиром и сушат в высоком вакууме. Остаток подвергают перекристаллизации из смеси 20 мл метанола с 20 мл этилацетата. Осадок вновь отсасывают на вакуум-фильтре, промывают этилацетатом и сушат в высоком вакууме. Получают 845 мг целевого соединения в виде бесцветных кристаллов (выход от теоретического 70%).
ВЭЖХ (метод 7): Rt=4,9 мин.
LC-MS (метод 10): Rt=1,62 мин; MS (ESIpos): m/z=735 (М+Н)+.
Пример 6
2-{4-[2-Амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}тио)-3,5-дицианопиридин-4-ил]фенокси}этил-N-(2-аминоэтил)-L-α-глутаминат дигидрохлорид
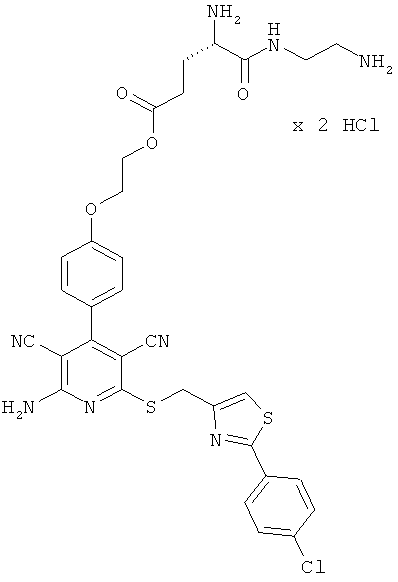
1 г (1,92 ммоль) 2-амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}тио)-4-[4-(2-гидроксиэтокси)фенил]пиридин-3,5-дикарбонитрила, 0,824 г (2,11 ммоль) соединения из примера 4А, 0,442 г (2,31 ммоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида, а также 0,024 г (0,19 ммоль) 4-диметиламинопиридина совмещают в смеси 40 мл дихлорметана с 10 мл ДМФ и в течение ночи перемешивают при комнатной температуре. Затем реакционные продукты выливают в смесь полунасыщенного раствора хлорида аммония с дихлорметаном. Органическую фазу отделяют, последовательно промывают водой, насыщенным раствором гидрокарбоната натрия и насыщенным раствором хлорида натрия, сушат над сульфатом магния, фильтруют и концентрируют. Остаток подвергают очистке флэш-хроматографией на силикагеле, используя в качестве элюента сначала смесь дихлорметана с этилацетатом (3:1), а затем смесь дихлорметан/этилацетат/метанол (градиент 300:100:5→300:100:10). Соответствующие фракции объединяют, растворитель удаляют отгонкой в вакууме. В результате сушки остатка в высоком вакууме получают 1,52 г защищенного промежуточного продукта (выход от теоретического 89%).
ВЭЖХ (метод 7): Rt=6,0 мин.
LC-MS (метод 11): Rt=2,54 мин; MS (ESIpos): m/z=891 (М+Н)+.
1,518 г (1,7 ммоль) промежуточного продукта вводят в 5 мл дихлорметана, смешивают с 5 мл безводной трифторуксусной кислоты, и реагенты в течение часа перемешивают при комнатной температуре. Реакционные продукты концентрируют, остаток промывают дихлорметаном. Затем остаток растворяют в 20 мл этилацетата. При перемешивании добавляют 20 мл 2М раствора водородхлорида в диэтиловом эфире. После кратковременного дополнительного перемешивания смесь отсасывают на вакуум-фильтре, остаток на фильтре промывают диэтиловым эфиром и сушат. Получают 1300 мг целевого соединения (выход от теоретического 99%). Продукт подвергают перекристаллизации из смеси 25 мл метанола с 25 мл этилацетата. Осадок вновь отсасывают на вакуум-фильтре, промывают этилацетатом и сушат в высоком вакууме. Получают 1080 мг целевого соединения (выход от теоретического 83%).
ВЭЖХ (метод 7): Rt=4,9 мин.
LC-MS (метод 10): Rt=1,59 мин; MS (ESIpos): m/z=691 (М+Н)+.
1Н-ЯМР (400 МГц, ДМСО-d6): δ=2,05 (m, 2Н), 2,45 (m, 2Н), 2,85 и 2,95 (2m, 2Н), 3,25 и 3,5 (2m, 2Н), 3,7 (m, 1Н), 4,3 (m, 2Н), 4,4 (m, 2Н), 4,65 (s, 2Н), 7,12 (d, 2Н), 7,48 (d, 2Н), 7,58 (d, 2Н), 7,93 (s, 1Н), 7,94 (d, 2Н), 8,1 (m, 3H), 8,45 (m, 3H), 8,95 (t, 1Н).
Пример 7
2-{4-[2-Амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}тио)-3,5-дицианопиридин-4-ил]фенокси}этил-N-(2-аминоэтил)-L-глутаминат дигидрохлорид
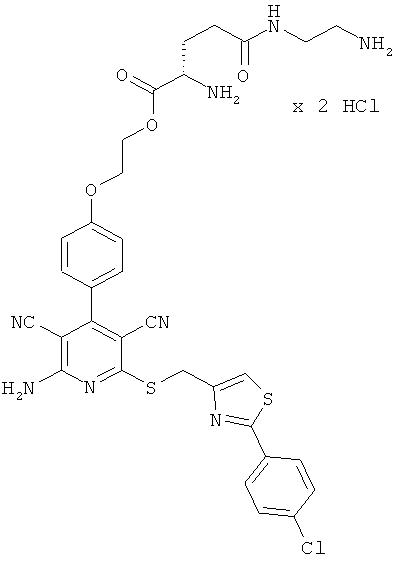
1 г (1,92 ммоль) 2-амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}тио)-4-[4-(2-гидроксиэтокси)фенил]пиридин-3,5-дикарбонитрила, 0,824 г (2,11 ммоль) соединения из примера 5А, 0,442 г (2,31 ммоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида, а также 0,024 г (0,192 ммоль) 4-диметиламинопиридина совмещают в смеси 40 мл дихлорметана с 10 мл ДМФ и в течение ночи перемешивают при комнатной температуре. Продукты реакции выливают в смесь полунасыщенного раствора хлорида аммония с дихлорметаном. Органическую фазу отделяют, последовательно промывают водой, насыщенным раствором гидрокарбоната натрия и насыщенным раствором хлорида натрия, сушат над сульфатом магния, фильтруют и концентрируют. Остаток выделяют из дихлорметана осаждением диэтиловым эфиром. Осадок отделяют фильтрованием, промывают диэтиловым эфиром и сушат в высоком вакууме. Получают 1,5 г защищенного промежуточного продукта (выход от теоретического 87%).
ВЭЖХ (метод 7): Rt=6,0 мин.
LC-MS (метод 10): Rt=3,12 мин; MS (ESIpos): m/z=891 (М+Н)+.
1,5 г (1,7 ммоль) промежуточного продукта вводят в 20 мл дихлорметана, смешивают с 5 мл безводной трифторуксусной кислоты, и реагенты в течение часа перемешивают при комнатной температуре. Реакционные продукты концентрируют, остаток несколько раз промывают толуолом. Затем остаток вводят в смесь 15 мл дихлорметана, 5 мл этилацетата и 1 мл метанола. При перемешивании добавляют 20 мл 2М раствора водородхлорида в диэтиловом эфире. После кратковременного дополнительного перемешивания смесь отсасывают на вакуум-фильтре, остаток на фильтре дважды промывают диэтиловым эфиром и сушат. Затем остаток растворяют в 50 мл разбавленной соляной кислоты (pH 3) и лиофилизуют. Получают 1265 мг целевого соединения (выход от теоретического 98%).
ВЭЖХ (метод 7): Rt=4,9 мин.
LC-MS (метод 11): Rt=1,19 мин; MS (ESIpos): m/z=691 (М+Н)+.
Пример 8
2-{4-[2-Амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}тио)-3,5-дицианопиридин-4-ил]фенокси}этил-N-(2-аминоэтил)-L-аспарагинат дигидрохлорид
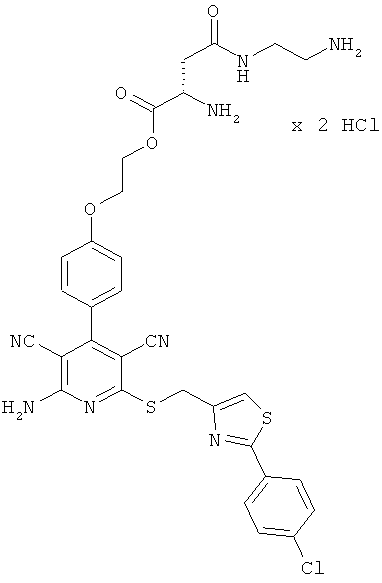
1 г (1,92 ммоль) 2-амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}тио)-4-[4-(2-гидроксиэтокси)фенил]пиридин-3,5-дикарбонитрила, 0,794 г (2,12 ммоль) соединения из примера 6А, 0,442 г (2,31 ммоль) 1-(3-диметил-аминопропил)-3-этилкарбодиимид гидрохлорида, а также 0,024 г (0,192 ммоль) 4-диметиламинопиридина совмещают в смеси 40 мл дихлорметана с 10 мл ДМФ и в течение ночи перемешивают при комнатной температуре. Затем продукты реакции выливают в смесь полунасыщенного раствора хлорида аммония с дихлорметаном. Органическую фазу отделяют, последовательно промывают водой, насыщенным раствором гидрокарбоната натрия и насыщенным раствором хлорида натрия, сушат над сульфатом магния, фильтруют и концентрируют. Остаток осаждают из дихлорметана диэтиловым эфиром. Осадок выделяют фильтрованием, промывают диэтиловым эфиром и сушат в высоком вакууме. Получают 1,28 г защищенного промежуточного продукта (выход от теоретического 74%).
ВЭЖХ (метод 7): Rt=6,0 мин.
LC-MS (метод 10): Rt=3,13 мин; MS (ESIpos): m/z=877 (М+Н)+.
1,28 г (1,46 ммоль) промежуточного продукта вводят в 20 мл дихлорметана, смешивают с 5 мл безводной трифторуксусной кислоты, и реагенты в течение часа перемешивают при комнатной температуре. Реакционные продукты концентрируют, и остаток несколько раз промывают толуолом. Затем остаток вводят в смесь 15 мл дихлорметана, 5 мл этилацетата и 1 мл метанола. При перемешивании добавляют 5 мл 2М раствора водородхлорида в диэтиловом эфире. После кратковременного дополнительного перемешивания смесь отсасывают на вакуум-фильтре, остаток на фильтре дважды промывают диэтиловым эфиром и сушат. Получают 1096 мг целевого соединения (количественный выход).
ВЭЖХ (метод 7): Rt=4,9 мин.
LC-MS (метод 10): Rt=1,58 мин; MS (ESIpos): m/z=677 (М+Н)+.
Пример 9
(2S)-5-(2-{4-[2-амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}тио)-3,5-дицианопиридин-4-ил]фенокси}этокси)-2-[(3-карбоксипропаноил)амино]-5-оксопентановая кислота
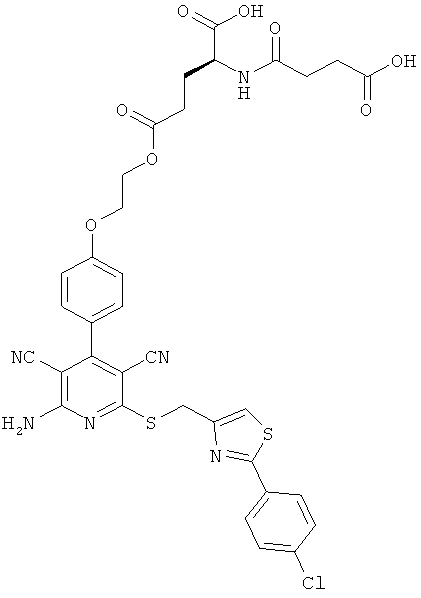
0,432 г (2,48 ммоль) 4-трет-бутокси-4-оксомасляной кислоты, 0,475 г (2,48 ммоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида, 0,380 г (2,48 ммоль) 1-гидрокси-1Н-бензотриазол гидрата, а также 1,59 г (2,254 ммоль) соединения из примера 7А совмещают в 70 мл ДМФ и в течение ночи перемешивают при комнатной температуре. Продукты реакции выливают в смесь полунасыщенного раствора хлорида аммония с этилацетатом. Органическую фазу отделяют, последовательно промывают водой, насыщенным раствором гидрокарбоната натрия и насыщенным раствором хлорида натрия, сушат над сульфатом магния, фильтруют и концентрируют. Остаток подвергают очистке флэш-хроматографией на силикагеле, используя в качестве элюента смесь дихлорметана с этилацетатом (градиент 10:1→5:1→3:1). Соответствующие фракции объединяют, и растворитель удаляют отгонкой в вакууме. Затем остаток отдельными порциями вновь подвергают очистке, используя препаративную ВЭЖХ (метод 6b). Соответствующие фракции объединяют, и растворитель удаляют отгонкой в вакууме. В результате сушки остатка в высоком вакууме получают 1,63 г защищенного промежуточного продукта (выход от теоретического 84%).
ВЭЖХ (метод 7): Rt=6,3 мин.
LC-MS (метод 11): Rt=2,71 мин.
MS (ESIpos): m/z=861 (М+Н)+.
1,285 г (1,49 ммоль) промежуточного продукта вводят в 10 мл дихлорметана, смешивают с 10 мл безводной трифторуксусной кислоты, и реагенты в течение часа перемешивают при комнатной температуре. Реакционные продукты концентрируют, остаток несколько раз промывают толуолом. Затем остаток смешивают с диэтиловым эфиром, образующееся твердое вещество отсасывают на вакуум-фильтре и промывают диэтиловым эфиром. В результате сушки остатка в высоком вакууме получают 1,06 г целевого соединения (выход от теоретического 95%).
ВЭЖХ (метод 7): Rt=5,4 мин.
LC-MS (метод 10): Rt=2,23 мин; MS (ESIpos): m/z=749 (М+Н)+.
Пример 10
Динатриевая соль (2S)-5-(2-{4-[2-амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}тио)-3,5-дицианопиридин-4-ил]фенокси}этокси)-2-[(3-карбокси-пропаноил)амино]-5-оксопентановой кислоты
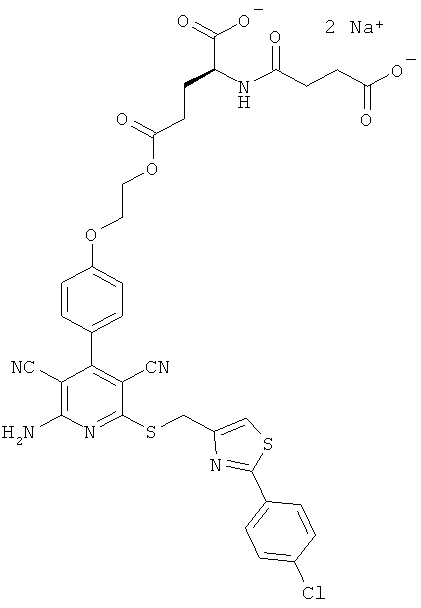
0,5 г (0,667 ммоль) соединения из примера 9 растворяют в смеси 12,5 мл ацетонитрила с 62,5 мл воды и смешивают с 13 мл 0,1 Н раствора едкого натра. После кратковременного перемешивания компоненты лиофилизуют. В результате сушки в высоком вакууме получают 0,53 г целевого соединения (количественный выход).
ВЭЖХ (метод 7): Rt=5,4 мин.
LC-MS (метод 11): Rt=2,06 мин; MS (ESIpos): m/z=749 (М+Н)+.
Пример 11
2-{4-[2-Амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}сульфанил)-3,5-дицианопиридин-4-ил]фенокси}этил-L-лизил-L-лейцинат дигидрохлорид
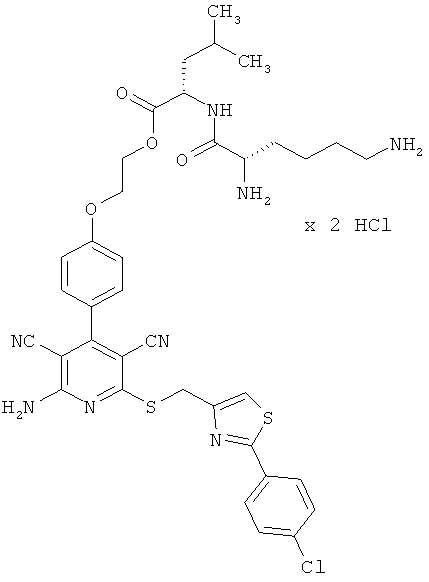
0,928 г (2,68 ммоль) N2,N6-бис(трет-бутоксикарбонил)-L-лизина, 0,362 г (2,68 ммоль) 1-гидрокси-1Н-бензотриазол гидрата и 0,411 г (2,144 ммоль) N-(3-диметиламинопропил)-N'-этилкарбодиимид гидрохлорида совмещают в 200 мл ДМФ. Добавляют 1,335 г (1,787 ммоль) соединения из примера 8А, а также 935 мкл N,N-диизопропилэтиламина, и компоненты в течение ночи перемешивают при комнатной температуре. Затем реакционную смесь концентрируют в вакууме, остаток вводят в этилацетат и последовательно экстрагируют водой, 5-процентной лимонной кислотой и дважды 5-процентным раствором гидрокарбоната натрия. Органическую фазу концентрируют, и остаток подвергают очистке флэш-хроматографией на силикагеле, используя в качестве элюента смесь дихлорметана с этилацетатом (градиент 3:1→2:1). Соответствующие фракции объединяют, растворитель удаляют отгонкой в вакууме. В результате сушки остатка в высоком вакууме получают 1,39 г защищенного промежуточного продукта (выход от теоретического 81%).
ВЭЖХ (метод 7): Rt=6,8 мин.
1,385 г (1,44 ммоль) полученного промежуточного продукта вводят в 20 мл дихлорметана и смешивают с 50 мл насыщенного раствора водородхлорида в дихлорметане. Продукты реакции в течение часа перемешивают при комнатной температуре, при этом целевой продукт выпадает в осадок. Растворитель удаляют выпариванием, остаток смешивают с 70 мл пентана, кратковременно перемешивают и отсасывают на вакуум-фильтре. В результате сушки остатка в высоком вакууме получают 1,17 г целевого соединения (выход от теоретического 97%).
ВЭЖХ (метод 7): Rt=5,17 мин.
LC-MS (метод 10): Rt=1,76 мин; MS (ESIpos): m/z=761 (М+Н)+.
Пример 12
2-{4-[2-Амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}сульфанил)-3,5-дицианопиридин-4-ил]фенокси}этил-L-лизил-D-аланинат дигидрохлорид
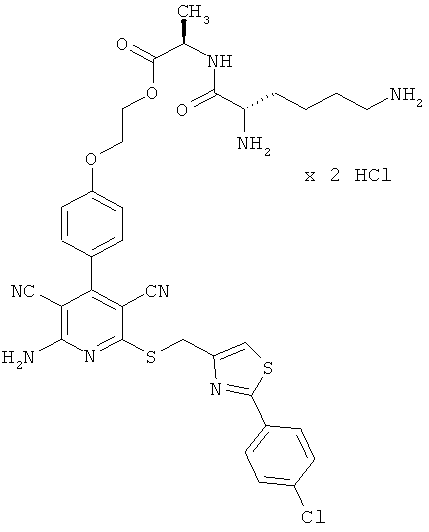
0,59 г (1,702 ммоль) N2,N6-бис(трет-бутоксикарбонил)-L-лизина, 0,345 г (2,553 ммоль) 1-гидрокси-1Н-бензотриазол гидрата и 0,391 г (2,042 ммоль) N-(3-диметиламинопропил)-N'-этилкарбодиимид гидрохлорида совмещают в 24 мл ДМФ. После 5-минутного перемешивания реагентов при комнатной температуре добавляют 1,2 г (1,702 ммоль) соединения из примера 9А, а также 1,5 мл N,N-диизопропилэтиламина, и продолжают перемешивание в течение ночи при комнатной температуре. Затем реакционную смесь концентрируют, и остаток распределяют между 500 мл этилацетата и 500 мл воды. Органическую фазу отделяют и последовательно экстрагируют трижды 5-процентной лимонной кислоты и трижды 10-процентным раствором гидрокарбоната натрия. Затем органическую фазу концентрируют, и остаток подвергают очистке флэш-хроматографией на силикагеле, используя в качестве элюента смесь дихлорметана с этилацетатом (градиент 2:1→1:1). Соответствующие фракции объединяют, растворитель удаляют отгонкой в вакууме. Остаток смешивают с 30 мл этилацетата, а затем с 100 мл диэтилового эфира. Смесь отсасывают на вакуум-фильтре, остаток на фильтре промывают диэтиловым эфиром. В результате сушки остатка в высоком вакууме получают 0,773 г защищенного промежуточного продукта (выход от теоретического 49%).
ВЭЖХ (метод 7): Rt=6,1 мин.
0,74 г (0,805 ммоль) полученного промежуточного продукта вводят в 200 мл дихлорметана. Через полученный раствор при перемешивании пропускают газообразный водородхлорид. При этом лишенное защиты целевое соединение выпадает в осадок. Продолжают перемешивание при комнатной температуре, и через час прекращают реакцию. Продукты реакции концентрируют в вакууме до половины от первоначального объема, и осадок выделяют фильтрованием. Остаток на фильтре промывают диэтиловым эфиром и сушат в высоком вакууме при 100°С. Получают 539 мг целевого соединения в виде бесцветных кристаллов (выход от теоретического 85%).
ВЭЖХ (метод 7): Rt=5,0 мин.
LC-MS (метод 13): Rt=1,04 мин; MS (ESIpos): m/z=719 (М+Н)+.
1Н-ЯМР (400 МГц, ДМСО-d6): δ=1,3 (d, 3H), 1,35 (m, 2Н), 1,52 (m, 2Н), 1,75 (m, 2Н), 2,75 (m, 2Н), 3,8 (m, 1Н), 4,3 (m, 2Н), 4,3-4,5 (m, 3H), 4,63 (s, 2Н), 7,12 (d, 2H), 7,48 (d, 2H), 7,58 (d, 2H), 7,8-8,0 (m, 5H), 8,25 (m, 2H), 9,0 (d, 1H).
Пример 13
2-{4-[2-Амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}сульфанил)-3,5-дицианопиридин-4-ил]фенокси}этил-N-(2-аминоэтил)-D-α-глутаминат дигидрохлорид
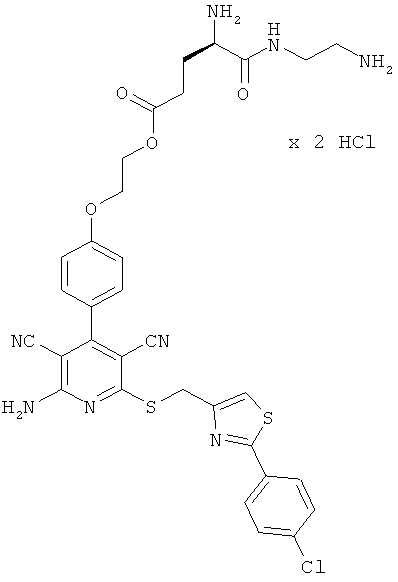
0,522 г (1,0 ммоль) 2-амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}-тио)-4-[4-(2-гидроксиэтокси)фенил]пиридин-3,5-дикарбонитрила, 0,430 г (1,104 ммоль) соединения из примера 10А, 0,231 г (1,204 ммоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида, а также 0,012 г (0,1 ммоль) 4-диметиламинопиридина совмещают в смеси 20 мл дихлорметана с 5 мл ДМФ и в течение ночи перемешивают при комнатной температуре. Продукты реакции выливают в смесь полунасыщенного раствора хлорида аммония с дихлорметаном. Органическую фазу отделяют, последовательно промывают водой, насыщенным раствором гидрокарбоната натрия и насыщенным раствором хлорида натрия, сушат над сульфатом магния, фильтруют и концентрируют. Остаток подвергают очистке флэш-хроматографией на силикагеле, используя в качестве элюента сначала смесь дихлорметана с этилацетатом (3:1), а затем смесь дихлорметан/этилацетат/метанол (градиент 300:100:5→300:100:10). Соответствующие фракции объединяют, растворитель удаляют отгонкой в вакууме. В результате сушки остатка в высоком вакууме получают 0,711 г защищенного промежуточного продукта (выход от теоретического 79%).
ВЭЖХ (метод 7): Rt=6,0 мин.
LC-MS (метод 13): Rt=1,54 мин; MS (ESIpos): m/z=891 (М+Н)+.
0,711 г (0,798 ммоль) полученного промежуточного продукта вводят в 4 мл дихлорметана, смешивают с 4 мл безводной трифторуксусной кислоты, и реагенты в течение часа перемешивают при комнатной температуре. Реакционные продукты концентрируют, остаток промывают дихлорметаном. Затем остаток растворяют в 50 мл этилацетата. При перемешивании добавляют 20 мл 2М раствора водородхлорида в диэтиловом эфире. После кратковременного дополнительного перемешивания смесь отсасывают на вакуум-фильтре, остаток на фильтре промывают диэтиловым эфиром и сушат. Получают 590 мг целевого соединения (выход от теоретического 97%).
ВЭЖХ (метод 7): Rt=4,9 мин.
LC-MS (метод 11): Rt=1,22 мин; MS (ESIpos): m/z=691 (М+Н)+.
1Н-ЯМР (400 МГц, ДМСО-d6): δ=2,05 (m, 2Н), 2,45 (m, 2Н), 2,85 и 2,95 (2m, 2Н), 3,25 и 3,5 (2m, 2Н), 3,7 (m, 1Н), 4,3 (m, 2Н), 4,4 (m, 2Н), 4,65 (s, 2Н), 7,12 (d, 2Н), 7,48 (d, 2Н), 7,58 (d, 2Н), 7,93 (s, 1Н), 7,94 (d, 2Н), 8,1 (m, 3H), 8,45 (m, 3H), 8,93 (t, 1Н).
Пример 14
2-{4-[2-Амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}сульфанил)-3,5-дицианопиридин-4-ил]фенокси}этил-L-аргинил-L-аланинат дигидрохлорид
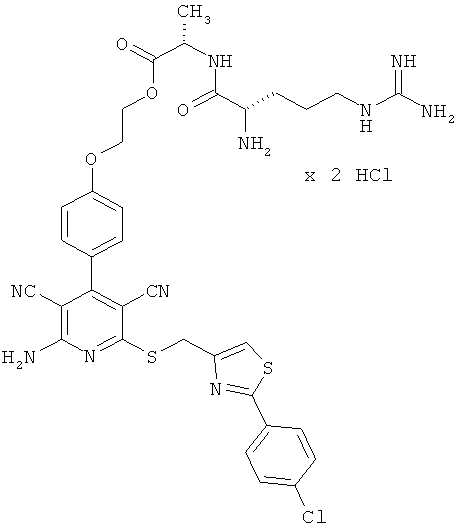
0,269 г (0,567 ммоль) N5-[N,N'-бис(трет-бутоксикарбонил)карбамидоил]-N2-(трет-бутоксикарбонил)-L-орнитина, 0,115 г (0,851 ммоль) 1-гидрокси-1Н-бензотриазол гидрата и 0,131 г (0,681 ммоль) N-(3-диметиламинопропил)-N'-этилкарбодиимид гидрохлорида совмещают в 20 мл ДМФ. После 30-минутного перемешивания добавляют 0,2 г (0,284 ммоль) соединения из примера 2А, а также 200 мкл N,N-диизопропилэтиламина, и компоненты в течение ночи перемешивают при комнатной температуре. Затем смесь концентрируют в вакууме, остаток вводят в дихлорметан и последовательно экстрагируют 5-процентной лимонной кислоты, 5-процентным раствором гидрокарбоната натрия и водой. Органическую фазу концентрируют, и остаток подвергают очистке флэш-хроматографией на силикагеле, используя в качестве элюента смесь дихлорметана с этилацетатом (3:1). Соответствующие фракции объединяют, растворитель удаляют отгонкой в вакууме. В результате сушки остатка в высоком вакууме получают 0,179 г защищенного промежуточного продукта (выход от теоретического 60%).
ВЭЖХ (метод 7): Rt=5,8 мин.
0,178 г (0,17 ммоль) полученного промежуточного продукта вводят в 30 мл насыщенного раствора водородхлорида в дихлорметане и в течение ночи перемешивают при комнатной температуре. Продукты реакции концентрируют в вакууме до половины от первоначального объема, и образовавшийся осадок выделяют фильтрованием. Остаток на фильтре промывают диэтиловым эфиром и сушат в высоком вакууме. Получают 119 мг целевого соединения в виде бесцветных кристаллов (выход от теоретического 82%).
ВЭЖХ (метод 7): Rt=4,9 мин.
LC-MS (метод 10): Rt=1,58 мин; MS (ESIpos): m/z=747 (М+Н)+.
1Н-ЯМР (400 МГц, ДМСО-d6): δ=1,35 (d, 3H), 1,55 (m, 2Н), 1,75 (m, 2Н), 3,25 (m, 2Н), 3,85 (m, 1Н), 4,25 (m, 2Н), 4,3-4,5 (m, 3H), 4,65 (s, 2Н), 7,12 (d, 2Н), 7,48 (d, 2Н), 7,58 (d, 2Н), 7,75 (t, 1Н), 7,93 (s, 1Н), 7,94 (d, 2Н), 8,3 (m, 3H), 9,1 (d, 1Н).
Пример 15
2-{4-[2-Амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}сульфанил)-3,5-дицианопиридин-4-ил]фенокси}этил-L-гистидил-L-аланинат дигидрохлорид
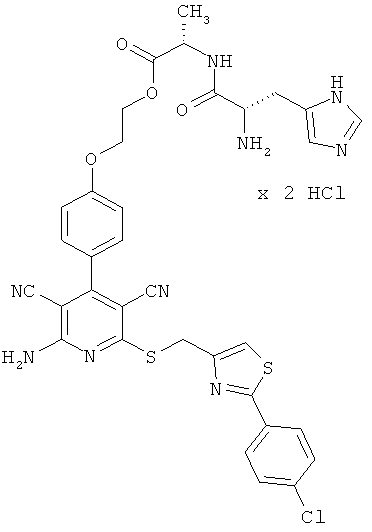
0,145 г (0,567 ммоль) N-(трет-бутоксикарбонил)-L-гистидина, 0,115 г (0,851 ммоль) 1-гидрокси-1Н-бензотриазол гидрата и 0,131 г (0,681 ммоль) N-(3-диметиламинопропил)-N'-этилкарбодиимид гидрохлорида совмещают в 20 мл ДМФ. После 30-минутного перемешивания добавляют 0,2 г (0,284 ммоль) соединения из примера 2А, а также 200 мкл N,N-диизопропилэтиламина, и компоненты в течение ночи перемешивают при комнатной температуре. Затем смесь концентрируют в вакууме, остаток вводят в дихлорметан и последовательно экстрагируют 5-процентной лимонной кислотой, 5-процентным раствором гидрокарбоната натрия и водой. Органическую фазу концентрируют, и остаток подвергают очистке флэш-хроматографией на силикагеле, используя в качестве элюента смесь толуола с этанолом (3:1). Соответствующие фракции объединяют, растворитель удаляют отгонкой в вакууме. В результате сушки остатка в высоком вакууме получают 0,206 г защищенного промежуточного продукта (выход от теоретического 88%).
ВЭЖХ (метод 7): Rt=5,3 мин.
0,206 г (0,249 ммоль) полученного промежуточного продукта вводят в 30 мл насыщенного раствора водородхлорида в дихлорметане и в течение двух часов перемешивают при комнатной температуре. Продукты реакции концентрируют в вакууме до половины от первоначального объема, и образовавшийся осадок выделяют фильтрованием. Остаток на фильтре промывают диэтиловым эфиром и сушат в высоком вакууме. Получают 171 мг целевого соединения в виде бесцветных кристаллов (выход от теоретического 90%).
ВЭЖХ (метод 7): Rt=4,8 мин.
LC-MS (метод 10): Rt=1,59 мин; MS (ESIpos): m/z=728 (М+Н)+.
1Н-ЯМР (400 МГц, ДМСО-d6): δ=1,35 (d, 3H), 3,15 и 3,25 (2dd, 2Н), 4,25 (m, 3H), 4,3-4,5 (m, 3H), 4,65 (s, 2Н), 7,12 (d, 2Н), 7,4 (s, 1Н), 7,48 (d, 2Н), 7,58 (d, 2Н), 7,93 (s, 1Н), 7,94 (d, 2Н), 8,5 (m, 3H), 9,05 (s, 1Н), 9,2 (d, 1Н).
Пример 16
2-{4-[2-Амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}сульфанил)-3,5-дицианопиридин-4-ил]фенокси}этил-N-[(2S)-2,4-диаминобутаноил]-L-аланинат дигидрохлорид
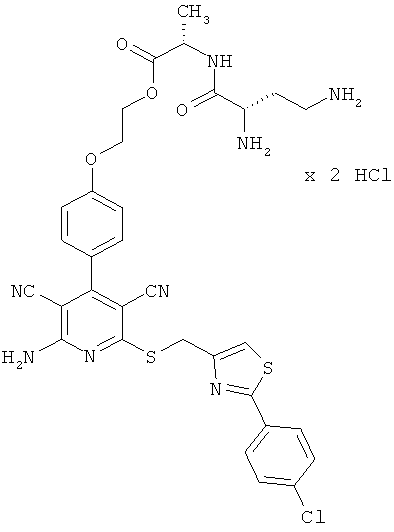
0,181 г (0,567 ммоль) (2S)-2,4-бис[(трет-бутоксикарбонил)амино]-масляной кислоты, 0,115 г (0,851 ммоль) 1-гидрокси-1Н-бензотриазол гидрата и 0,131 г (0,681 ммоль) N-(3-диметиламинопропил)-N'-этилкарбодиимид гидрохлорида совмещают в 20 мл ДМФ. После 30-минутного перемешивания при комнатной температуре добавляют 0,2 г (0,284 ммоль) соединения из примера 2А, а также 200 мкл N,N-диизопропилэтиламина, и компоненты в течение ночи перемешивают при комнатной температуре. Затем смесь концентрируют в вакууме, остаток вводят в дихлорметан и последовательно экстрагируют 5-процентной лимонной кислоты, 5-процентным раствором гидрокарбоната натрия и водой. Органическую фазу концентрируют, остаток подвергают очистке флэш-хроматографией на силикагеле, используя в качестве элюента смесь толуола с этанолом (10:1). Соответствующие фракции объединяют, растворитель удаляют отгонкой в вакууме. В результате сушки остатка в высоком вакууме получают 0,217 г защищенного промежуточного продукта (выход от теоретического 86%).
ВЭЖХ (метод 7): Rt=6,3 мин.
0,212 г (0,238 ммоль) полученного промежуточного продукта вводят в 25 мл насыщенного раствора водородхлорида в дихлорметане и в течение 1,5 часов перемешивают при комнатной температуре. Продукты реакции концентрируют в вакууме до половины от первоначального объема, и образовавшийся осадок выделяют фильтрованием. Остаток на фильтре промывают диэтиловым эфиром и сушат в высоком вакууме. Получают 171 мг целевого соединения в виде бесцветных кристаллов (выход от теоретического 94%).
ВЭЖХ (метод 7): Rt=4,8 мин.
LC-MS (метод 10): Rt=1,56 мин; MS (ESIpos): m/z=691 (М+Н)+.
Пример 17
2-{4-[2-Амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}сульфанил)-3,5-дицианопиридин-4-ил]фенокси}этил-L-лизилглицинат дигидрохлорид
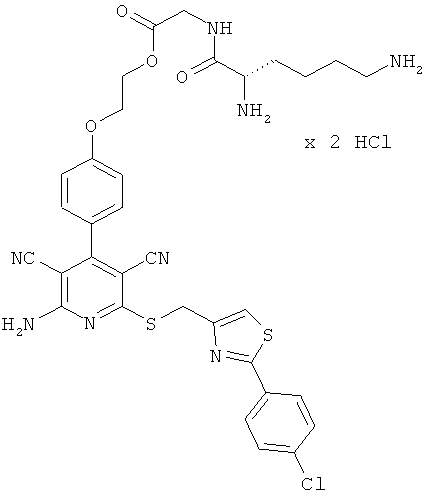
0,185 г (0,535 ммоль) N2,N6-бис(трет-бутоксикарбонил)-L-лизина, 0,108 г (0,803 ммоль) 1-гидрокси-1Н-бензотриазол гидрата и 0,123 г (0,642 ммоль) N-(3-диметиламинопропил)-N'-этилкарбодиимид гидрохлорида совмещают в 15 мл ДМФ и в течение 5 минут перемешивают при комнатной температуре. Затем добавляют 0,37 г (0,535 ммоль) соединения из примера 11А, а также 466 мкл N,N-диизопропилэтиламина, и компоненты в течение ночи перемешивают при комнатной температуре. Реакционные продукты концентрируют в вакууме, остаток распределяют между 500 мл этилацетата и 200 мл воды. Органическую фазу отделяют, последовательно экстрагируют трижды 5-процентной лимонной кислотой и трижды 5-процентным раствором гидрокарбоната натрия и сушат над сульфатом магния. Органическую фазу концентрируют, остаток в течение 10 мин перемешивают с 50 мл этилацетата. Затем медленно добавляют 100 мл диэтилового эфира, и образовавшийся осадок отсасывают на вакуум-фильтре. В результате сушки в высоком вакууме получают 0,303 г защищенного промежуточного продукта (выход от теоретического 63%).
ВЭЖХ (метод 7): Rt=6,1 мин.
0,279 г (0,308 ммоль) полученного промежуточного продукта вводят в 120 мл дихлорметана. Через полученный раствор при перемешивании пропускают газообразный водородхлорид, при этом лишенное защиты целевое соединение выпадает в осадок. Продукты реакции в течение последующего часа перемешивают при комнатной температуре. Затем смесь концентрируют в вакууме до половины от первоначального объема и медленно добавляют 30 мл абсолютного ТГФ. Смесь перемешивают в течение последующих 15 минут, образующийся осадок выделяют фильтрованием. Остаток на фильтре промывают диэтиловым эфиром и сушат в высоком вакууме. Получают 212 мг целевого соединения в виде бесцветных кристаллов (выход от теоретического 88%).
ВЭЖХ (метод 7): Rt=4,8 мин.
LC-MS (метод 10): Rt=1,52 мин; MS (ESIpos): m/z=705 (M+H)+.
Пример 18
2-{4-[2-Амино-6-({[2-(4-хлорфенил)-1,3-тиазол-4-ил]метил}сульфанил)-3,5-дицианопиридин-4-ил]фенокси}этил-L-лизил-L-фенилаланинат дигидрохлорид
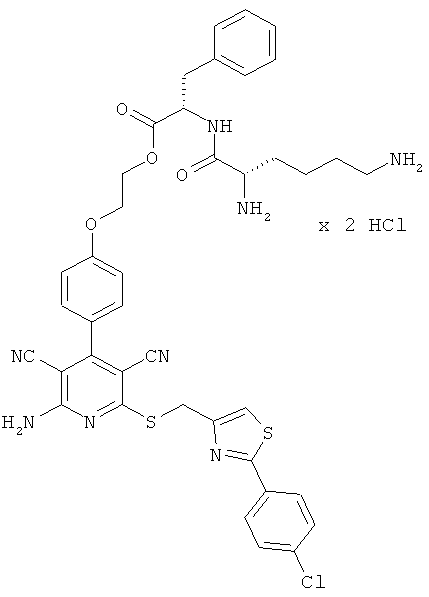
0,166 г (0,48 ммоль) N2,N6-бис(трет-бутоксикарбонил)-L-лизина, 0,097 г (0,72 ммоль) 1-гидрокси-1Н-бензотриазол гидрата и 0,110 г (0,576 ммоль) N-(3-диметиламинопропил)-N'-этилкарбодиимид гидрохлорида совмещают в 7 мл ДМФ и в течение 5 минут перемешивают при комнатной температуре. Затем добавляют 0,375 г (0,48 ммоль) соединения из примера 12А, а также 418 мкл N,N-диизопропилэтиламина, и компоненты в течение ночи перемешивают при комнатной температуре. Реакционные продукты концентрируют в вакууме, остаток вводят в 200 мл этилацетата, последовательно экстрагируют дважды 10-процентной лимонной кислотой и дважды 10-процентным раствором гидрокарбоната натрия и сушат над сульфатом магния. Органическую фазу концентрируют, остаток вводят в 10 мл этилацетата, и целевой продукт осаждают путем добавления диэтилового эфира. Осадок отсасывают на вакуум-фильтре и сушат в высоком вакууме. Получают 0,338 г защищенного промежуточного продукта (выход от теоретического 71%).
ВЭЖХ (метод 7): Rt=6,7 мин.
0,32 г (0,321 ммоль) полученного промежуточного продукта вводят в 100 мл дихлорметана. Через полученный раствор при перемешивании пропускают газообразный водородхлорид, при этом лишенное защиты целевое соединение выпадает в осадок. Продукты реакции в течение последующего часа перемешивают при комнатной температуре, концентрируют в вакууме до половины от первоначального объема и медленно добавляют 10 мл абсолютного ТГФ. Смесь перемешивают в течение последующих 15 минут, образующийся осадок выделяют фильтрованием. Остаток на фильтре промывают диэтиловым эфиром и сушат в высоком вакууме. Получают 188 мг целевого соединения в виде бесцветных кристаллов (выход от теоретического 67%).
ВЭЖХ (метод 7): Rt=5,0 мин.
LC-MS (метод 11): Rt=1,33 мин; MS (ESIpos): m/z=795 (M+H)+.
В. Определение растворимости, стабильности и способности к высвобождению
а) Определение растворимости
Испытуемое соединение суспендируют в 5-процентном водном растворе декстрозы. Полученную суспензию в течение 24 часов встряхивают при комнатной температуре. После 30-минутного ультрацентрифугирования с ускорением 224000g надосадочную жидкость разбавляют ДМСО и анализируют методом ВЭЖХ. Количественную оценку результатов анализа выполняют на основании двухточечной калибровочной кривой испытуемого соединения в ДМСО.
Анализ кислот методом ВЭЖХ
Прибор Agilent 1100 с детектором DAD (G1315A), четырехходовым насосом (G1311A), автоматической пипеткой (СТС HTS PAL), дегазатором (G1322A) и термостатом колонки (G1316A); колонка: Phenomenex Gemini С18, 5 мкм, 50 мм × 2 мм; температура 40°С; элюент А: вода/фосфорная кислота с pH 2, элюент В: ацетонитрил; расход 0,7 мл/мин; градиент: 0-0,5 мин 85% А, 15% В; линейно 0,5-3 мин 10% А, 90% В; 3-3,5 мин 10% А, 90% В; линейно 3,5-4 мин 85% А, 15% В; 4-5 мин 85% А, 15% В.
Анализ оснований методом ВЭЖХ
Прибор Agilent 1100 с детектором DAD (G1315A), четырехходовым насосом (G1311A), автоматической пипеткой (СТС HTS PAL), дегазатором (G1322A) и термостатом колонки (G1316A); колонка: VDSoptilab Kromasil 100 С18, 3,5 мкм, 60 мм × 2,1 мм; температура 30°С; элюент А: вода + 5 мл перхлорной кислоты в литре, элюент В: ацетонитрил; расход: 0,75 мл/мин; градиент: 0-0,5 мин 98% А, 2% В; линейно 0,5-4,5 мин 10% А, 90% В; 4,5-6 мин 10% А, 90% В; линейно 6,5-6,7 мин 98% А, 2% В; 6,7-7,5 мин 98% А, 2% В.
В таблице 1 приведена растворимость соединений из репрезентативных примеров осуществления изобретения в 5-процентном водном растворе декстрозы
|
Деструкция соединений в указанном растворе отсутствует.
Растворимость лежащего в основе испытуемых соединений действующего вещества [соединения формулы (А)] в 5-процентном водном растворе декстрозы составляет менее 0,1 мг/литр.
b) Стабильность в буферных растворах при варьируемых значениях показателя pH
В пробирке для ВЭЖХ объемом 2 мл берут навеску испытуемого соединения (0,3 мг) и смешивают ее с 0,5 мл смеси ацетонитрил/ДМСО (9:1). С целью растворения испытуемого соединения пробирку примерно на 10 секунд помещают в ультразвуковую ванну. Затем добавляют 0,5 мл соответствующего (буферного) раствора, и образец вновь подвергают ультразвуковой обработке.
Используемые буферные растворы
|
В течение 24 часов при 25°С ежечасно выполняют ВЭЖХ-анализ образцов раствора объемом 5 мкл на содержание остающегося без изменений испытуемого соединения, соответственно образующегося исходного вещества формулы (А). Количественную оценку результатов анализа выполняют на основании выраженных в процентах площадей соответствующих пиков.
Метод ВЭЖХ
Прибор Agilent 1100 с детектором DAD (G1314A), бинарным насосом (G1312A), автоматической пипеткой (G1329A), печью (G1316A) и термостатом (G1330A); колонка Kromasil 100 С18, 125 мм × 4 мм, 5 мкм; температура колонки 30°С; элюент А: вода + 5 мл перхлорной кислоты в литре, элюент В: ацетонитрил; градиент: 0-2,0 мин 90% А, 10% В; 2,0-18,0 мин 64% А, 36% В; 18,0-20,0 мин 64% А, 36% В; 20,0-21,0 мин 10% А, 90% В; 21,0-23,0 мин 90% А, 10% В; 23,0-26,0 мин 90% А, 10% В; расход 2,0 мл/мин; УФ-детектирование 294 нм.
В таблице 2 приведено содержание испытуемых соединений, полученных в репрезентативных примерах осуществления изобретения, определенное в виде отношений площадей пиков (F) в соответствующие моменты времени (t) к их площадям в начальный момент времени.
|
В условиях данного испытания одновременно со снижением содержания испытуемого соединения констатируют рост содержания действующего вещества [соединения формулы (А)].
с) Стабильность в крысиной и человеческой плазме in vitro
В пробирке для ВЭЖХ объемом 2 мл берут навеску испытуемого соединения (1 мг) и смешивают ее с 1,5 мл ДМСО и 1 мл воды. С целью растворения испытуемого соединения пробирку примерно на 10 секунд помещают в ультразвуковую ванну. К 0,5 мл соответствующего раствора добавляют 0,5 мл крысиной, соответственно человеческой плазмы с температурой 37°С. Пробирку встряхивают и отбирают на анализ первый образец объемом около 10 мкл (момент времени t0). В течение промежутка времени после начала инкубации, составляющего до 2 часов, отбирают от 4 до 6 предназначенных для количественной оценки дополнительных аликвотных образцов. Образцы во время испытания выдерживают при температуре 37°С. Определение характеристик и количественную оценку выполняют методом ВЭЖХ.
Метод ВЭЖХ
Прибор Agilent 1100 с детектором DAD (G1314A), бинарным насосом (G1312A), автоматической пипеткой (G1329A), печью (G1316A) и термостатом колонки (G1330A); колонка Kromasil 100 С18, 250 мм × 4 мм, 5 мкм; температура колонки 30°С; элюент А: вода + 5 мл перхлорной кислоты в литре, элюент В: ацетонитрил; градиент: 0-8,0 мин 53% А, 47% В; 8,0-18,0 мин 53% А, 47% В; 18,0-20,0 мин 90% А, 10% В; 20,0-21,0 мин 90% А, 10% В; 21,0-22,5 мин 98% А, 2% В; 22,5-25,0 мин 98% А, 2% В; расход 2 мл/мин; УФ-детектирование 294 нм.
В таблице 3 приведены значения времени (t50%A), при которых после совместной инкубации полученных в репрезентативных примерах испытуемых соединений с крысиной плазмой образуется 50% от максимально возможного количества действующего вещества [соединения формулы (А)]. Количественную оценку выполняют по отношению площадей пиков в соответствующие моменты времени и в начальный момент инкубации.
|
d) Исследование фармакокинетики на крысах Wistar при внутривенной инъекции испытуемых соединений
В день применения испытуемых соединений в яремную вену наркотизированных посредством Isofluran® подопытных животных (мужских особей крыс Wistar с массой тела 200-250 г) имплантируют используемый при последующем отборе крови катетер.
В день эксперимента в хвостовую вену крысы стеклянным шприцем Hamilton® вводят определенную дозу испытуемого соединения в виде соответствующего раствора (длительность применения менее 10 секунд). В течение последующих 24 часов посредством катетера последовательно отбирают от 8 до 12 образцов крови. С целью выделения плазмы образцы центрифугируют в гепаринизованных трубочках. С целью осаждения белка определенный объем плазмы в соответствующий момент времени смешивают с ацетонитрилом. После центрифугирования, используя соответствующий метод LC/MS-MS, количественно определяют содержание испытуемого соединения и при необходимости известных продуктов его деструкции в надосадочной жидкости.
На основании измеренных концентраций в плазме вычисляют фармакокинетические характеристики испытуемого соединения, соответственно соединения формулы (А), высвобождающегося из испытуемого соединения в качестве действующего вещества, такие как AUC, Cmax, Т1/2 (период полувыделения) и CL (коэффициент очищения крови).
После внутривенного применения соединений из примеров 4, 5, 6 и 7 их не удавалось обнаружить в плазме уже при первом анализе крови. В течение всего периода испытания (24 часа) в плазме обнаруживали только действующее вещество формулы (А).
е) Исследование фармакокинетики на крысах Wistar при оральном применении испытуемых соединений
В день применения испытуемых соединений в яремную вену наркотизированных посредством Isofluran® подопытных животных (мужских особей крыс Wistar с массой тела 200-250 г) имплантируют используемый при последующем отборе крови катетер.
В день эксперимента в желудок животного через желудочный зонд вводят определенную дозу испытуемого соединения в виде соответствующего раствора. В течение последующих 24 часов посредством катетера последовательно отбирают от 8 до 12 образцов крови. С целью выделения плазмы образцы центрифугируют в гепаринизованных трубочках. С целью осаждения белка определенный объем плазмы в соответствующий момент времени смешивают с ацетонитрилом. После центрифугирования, используя соответствующий метод LC/MS-MS, количественно определяют содержание испытуемого соединения и при необходимости известных продуктов его деструкции в надосадочной жидкости.
На основании измеренных концентраций в плазме вычисляют фармакокинетические характеристики испытуемого соединения, соответственно соединения формулы (А), высвобождающегося из испытуемого соединения в качестве действующего вещества, такие как AUC, Cmax, Т1/2 (период полувыделения) и CL (коэффициент очищения крови).
После орального применения соединений из примеров 4, 5 и 6 их не удавалось обнаружить в плазме уже при первом анализе крови. В течение всего периода испытания (24 часа) в плазме обнаруживали только действующее вещество формулы (А).
f) Влияние испытуемых соединений на частоту сердечных сокращений находящихся под наркозом крыс
Используют мужские особи крыс Wistar с массой тела более 250 г. В ночь перед экспериментом животным не дают корм, однако они имеют свободный доступ к питьевой воде. Животных препарируют и исследуют под наркозом, выполняемым посредством Trapanal® (100 мг/кг массы тела). Инъекцию и инфузию осуществляют через катетер в яремной вене, кровяное давление регистрируют посредством катетера в бедренной вене (трансдуцер фирмы Braun, Мелсунген). С целью компенсации потерь жидкости после препарирования выполняют длительное внутривенное вливание физиологического раствора поваренной соли. Испытуемое соединение или раствор плацебо применяют внутривенно по истечении времени уравновешивания (около 1 часа) в виде ударной дозы. Частоту сердечных сокращений и артериальное кровяное давление во время уравновешивания и по истечении по меньшей мере 30 минут после ударной инъекции записывают с помощью цифровой программы анализа.
В таблице 4 приведено снижение максимальной частоты сердечных сокращений в первые 30 минут после ударной внутривенной инъекции (100 мкг/кг) действующего вещества формулы (А), соответственно эквивалентной дозировки соединений из репрезентативных примеров осуществления изобретения.
|
С. Примеры изготовления фармацевтических препаратов
Предлагаемые в изобретении соединения могут быть преобразованы, например, в следующие фармацевтические препараты.
Таблетки
Состав
100 мг предлагаемого в изобретении соединения, 50 мг лактозы в виде моногидрата, 50 мг природного кукурузного крахмала, 10 мг
поливинилпирролидона PVP 25 фирмы BASF (Людвигсхафен, Германия) и 2 мг стерата магния.
Масса таблетки 212 мг. Диаметр 8 мм, радиус кривизны 12 мм.
Изготовление
Смесь предлагаемого в изобретении соединения, лактозы и крахмала гранулируют, используя раствор поливинилпирролидона в воде концентрацией 5% мас. Гранулят после сушки смешивают в течение 5 минут со стератом магния. Полученную смесь подвергают прессованию на обычном таблеточном прессе (параметры таблеток указан выше). Нормированное усилие прессования составляет 15 кН.
Орально применяемая суспензия
Состав
1000 мг предлагаемого в изобретении соединения, 1000 мг этанола (96%), 400 мг ксантановой смолы Rhodigel® фирмы FMC (США, Пенсильвания) и 99 г воды.
Однократной дозе, содержащей 100 мг предлагаемого в изобретении соединения, соответствует 10 мл орально применяемой суспензии.
Изготовление
Смолу Rhodigel® суспендируют в этаноле и к полученной суспензии добавляют предлагаемое в изобретении соединение. При перемешивании добавляют воду. Компоненты перемешивают около 6 часов до завершения набухания смолы Rhodigel®.
Орально применяемый раствор
Состав
500 мг предлагаемого в изобретении соединения, 2,5 г полисорбата и 97 г полиэтиленгликоля ПЭГ 400. Однократной дозе, содержащей 100 мг предлагаемого в изобретении соединения, соответствует 20 мл орально применяемого раствора.
Изготовление
Предлагаемое в изобретении соединение при перемешивании суспендируют в смеси полиэтиленгликоля с полисорбатом. Перемешивание продолжают до полного растворения предлагаемого в изобретении соединения.
Раствор для внутривенных инъекций
Предлагаемое в изобретении соединение в концентрации ниже предела насыщения растворяют в физиологически совместимом растворителе (например, в изотоническом растворе хлорида натрия, 5-процентном растворе глюкозы и/или 30-процентном растворе полиэтиленгликоля ПЭГ 400, показатель pH которых устанавливают в диапазоне от 3 до 5). Раствор фильтруют при необходимости в стерильных условиях и/или расфасовывают в стерилизованные и освобожденные от пирогенных примесей сосуды для инъекций.
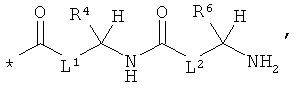
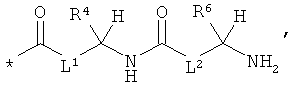
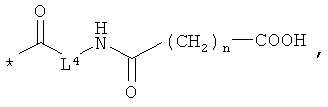
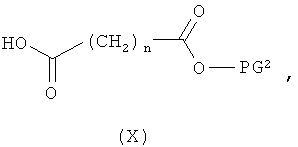
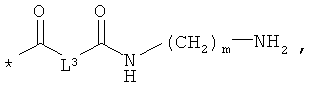
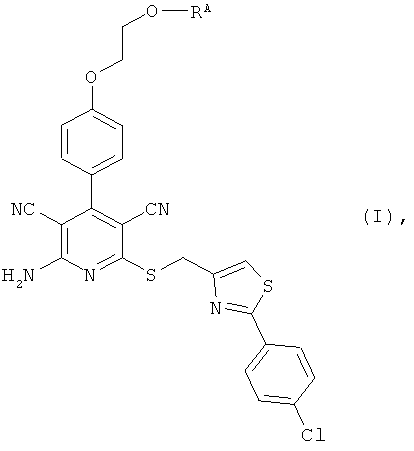
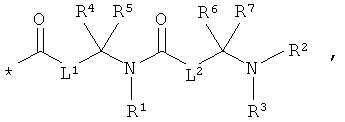
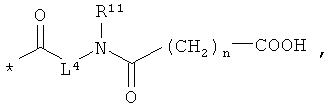
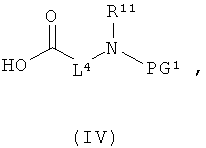
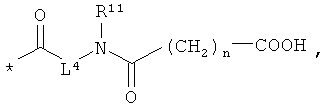
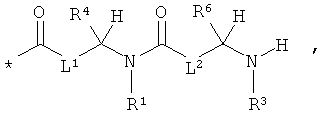
Комбинированный продукт для борьбы с паразитами животных
Трансдермальное применение триазинов для борьбы с инфекциями кокцидий
Способ изготовления твердого, орально применимого фармацевтического состава
4-(4-циано-2-тиоарил)-дигидропиримидиноны и их применение
Шнековые элементы для экструзии пластических масс
Составы, содержащие триазиноны и железо
Замещенные дигидропиразолоны в качестве ингибиторов hif-пролил-4-гидроксилазы
Способ изготовления шнековых элементов
Шнековые элементы с улучшенной эффективностью диспергирования и уменьшенным поступлением энергии
Шнековые элементы со сниженной энергоподачей при повышении давления
Комбинированный продукт для борьбы с паразитами животных
Трансдермальное применение триазинов для борьбы с инфекциями кокцидий
Устойчивый к воздействию температуры катализатор для окисления хлороводорода в газовой фазе
Способ изготовления твердого, орально применимого фармацевтического состава
Элемент безопасности
Оксазолидиноны для лечения и/или профилактики расстройств сердечной деятельности
4-(4-циано-2-тиоарил)-дигидропиримидиноны и их применение
Шнековые элементы для экструзии пластических масс
Составы, содержащие триазиноны и железо
Замещенные дигидропиразолоны в качестве ингибиторов hif-пролил-4-гидроксилазы

