Результат интеллектуальной деятельности: СПОСОБ ОПРЕДЕЛЕНИЯ ПРОИЗВОДНЫХ НИТРОФУРАНА, ПИРАЗОЛА, ИЗОНИКОТИНОВОЙ КИСЛОТЫ, ТИОАМИНОКИСЛОТ В ЛЕКАРСТВЕННЫХ ФОРМАХ
Вид РИД
Изобретение
Изобретение относится к химическим способам анализа, в частности к определению производных нитрофурана, пиразола, изоникотиновой кислоты, тиоаминокислот в лекарственных формах.
Одним из важнейших факторов, определяющих качество лекарственных средств, изготовляемых в аптеках, являются постановка и выполнение внутриаптечного контроля. По мере развития аптечного дела и совершенствования лекарственной помощи населению расширился ассортимент лекарственных веществ, усложнились состав и химический анализ лекарственных средств, изготовляемых в аптеках. Лекарственные формы, как правило, содержат 3-4 и более веществ из различных групп химических соединений, для разделения, идентификации и количественного определения которых необходимы быстро выполняемые и надежные методики анализа [Кулешова М.И. Анализ лекарственных форм, изготовляемых в аптеках / М.И.Кулешова, Л.Н.Гусева, O.K.Сивицкая; М.: Медицина, 1989, с.3-7].
Контроль за качеством лекарственных препаратов и установления сроков годности невозможен без широких количественных методов анализа [Государственная фармакопея. Вып.1. / Бабков Ю.Г., Бабаян Э.А., Машковский М.Д., Обоймакова А.Р., Булаев В.М., Гуськова Л.С., Лепахин В.К., Любимов Б.И., Натрадзе А.Г., Соколов С.Д., Тенцова А.И.; М.: Медицина, 1987. - 335 с.; Государственная фармакопея. Вып.2. / Бабков Ю.Г., Бабаян Э.А., Машковский М.Д., Обоймакова А.Р., Булаев В.М., Гуськова Л.С., Лепахин В.К., Любимов Б.И., Натрадзе А.Г., Соколов С.Д., Тенцова А.И.; М.: Медицина, 1990. - 385 с].
Известен способ определения количественного содержания примеси 4-метиламиноантипирина в многокомпонентных лекарственных препаратах жаропонижающего, аналгезирующего, противопростудного действия, в котором анализ проводили методом высокоэффективной жидкостной хроматографии (ВЭЖХ) с обращенно-фазовой хроматографической колонкой и ультрафиолетовым (УФ) детектором. В качестве подвижных фаз (ПФ) использовали растворы, содержащие ацетонитрил, 0,025 М раствор калия фосфорнокислого однозамещенного и воду (с различными объемными отношениями). Детектирование проводили при 244 нм. Получали не менее 3 хроматограмм (RU №2338189 МПК G01N 33/15, G01N 30/00, G01N 30/36 10.11.2008).
Недостатком описанного способа является длительность исполнения.
Известен способ одновременного количественного определения состава многокомпонентных лекарственных препаратов методом обращенно-фазовой ВЭЖХ с помощью УФ-детектора. При этом состав ПФ изменялся от воды к фосфатному буферному раствору с рН 3,0 до смеси его с ацетонитрилом в объемном отношении 1:1, получали не менее 3 хроматограмм каждого раствора (RU №2267115 7 МПК G01N 21/33, G01N 30/36, G01N 30/34, 27.12.2005).
Недостатком способа является сложность предварительной пробоподготовки и длительность выполнения анализа.
Известен способ определения количественного состава таблеток «Пеналгин ФС», в котором анализ проводили методом высокоэффективной жидкостной хроматографии (ВЭЖХ) с обращенно-фазовой хроматографической колонкой и ультрафиолетовым (УФ) детектором. Анализ проводили в обращенно-фазовом изократическом режиме при концентрациях ацетонитрила и калия фосфата однозамещенного в подвижной фазе 4÷6% (по объему) и 0,09÷0,12% (весо-объемных) соответственно (RU №2332662 7 МПК G01N 33/15, 27.08.2008).
Недостатком способа является отсутствие зависимости удерживания разнообразных классов веществ от концентрации ацетонитрила и калия фосфата однозамещенного в подвижной фазе, а также длительности исполнения анализа.
Известен способ определения чистоты фтивазида и метизида, основанного на хроматографировании пластинки в УФ-свете. Спиртовые растворы испытуемого лекарственного вещества наносили на хроматографическую пластинку «Армсорб» или «Сорбфил», после высушивания хроматографировали восходящим методом в системе растворителей хлороформ - спирт 95% - ледяная уксусная кислота - 25%-ный раствор аммиака в соотношении 9:1:1:0,5 (RU №2225205, 7 МПК А61К 31/4409, А61К 31/455, А61Р 31/06).
Недостатком описанного способа является его многостадийность и, как следствие, трудоемкость, длительность исполнения.
Известен способ определения производных пиразолона (анальгин, антипирин), основанный на взаимодействии препарата или продукта его гидролиза при определенном значении рН с раствором йода. Во избежание обратимости реакции йодистоводородную кислоту нейтрализуют ацетатом натрия. Необходимо также иметь в виду, что йодопирин адсорбирует на своей поверхности йод, вследствие чего для растворения осадка прибавляют хлороформ [Методы анализа лекарств / Максютина Н.П., Каган Ф.Е., Кириченко Л.А., Митченко Ф.А.; Киев: Здоровья, 1984].
Известен йодометрический способ определения производных нитрофурана (фурацилин), основанный на окислении гидразиновой группировки раствором йода в щелочной среде [Кулешова М.И. Анализ лекарственных форм, изготовляемых в аптеках / М.И.Кулешова, Л.Н.Гусева, O.K.Сивицкая; М.: Медицина, 1989, с.167-168].
Известен йодометрический способ определения производных изоникотиновой кислоты (фтивазида), основанный на окислении препарата раствором йода в гидрокарбонатной среде [Перельман Я.М. Анализ лекарственных форм / Я.М.Перельман. - Л.: Медгиз, 1961, с.294-296].
Известен йодометрический способ определения тиоаминокислот (метионин), основанный на окислении серы в α-амино-γ-метилтиомасляной кислоте до четырехвалентной раствором йода, избыток которого оттитровывают раствором тиосульфата натрия [Кулешова М.И. Анализ лекарственных форм, изготовляемых в аптеках / М.И.Кулешова, Л.Н.Гусева, O.К.Сивицкая; М.: Медицина, 1989, с.177-178].
Наиболее близким к заявленному изобретению является способ йодометрического определения состава фармацевтических препаратов: анальгина, фурацилина, метионина, фтивазида, антипирина, заключающийся в переводе препарата в анализируемую форму, прибавлении к пробе титрованного избытка раствора йода при определенном значении рН и оттитровывании избытка йода раствором тиосульфата натрия, с индикаторным (крахмал) или безиндикаторным фиксированием точки эквивалентности. Все эти способы представлены в Государственной фармакопее СССР и имеют законодательный характер [Государственная фармакопея СССР / Бабков Ю.Г., Бабаян Э.А., Машковский М.Д., Обоймакова А.Р., Булаев В.М., Гуськова Л.С., Лепахин В.К., Любимов Б.И., Натрадзе А.Г., Соколов С.Д., Тенцова А.И.; М.: Медицина. 1987. С.173-175]. Для анализа может быть использован метод прямого и обратного титрования.
Недостатком этих способов йодометрического определения составов является низкая чувствительность определения (так как имеем дело с классической титриметрией), длительность выполнения анализа, летучесть самого титранта, влияние кислотности на результаты определения и наличие индикаторной ошибки.
Задачей настоящего изобретения является разработка экспрессного и достоверного способа определения производных нитрофурана, пиразола, изоникотиновой кислоты и аминокислот в лекарственных формах.
Технический результат заявленного изобретения состоит в упрощении способа, уменьшении продолжительности определения, а также повышении точности и предела обнаружения органических веществ в фармацевтических препаратах.
Это достигается тем, что способ определения производных нитрофурана, пиразола, изоникотиновой кислоты, тиоаминокислот в лекарственных формах, заключающийся в предварительном переводе анализируемого препарата в жидкую форму, помещение его в ячейку, содержащую определенное количество генерированного йода, полученного путем облучения стабилизированным источником света реакционной смеси, состоящей из 0,5М раствора йодида калия, буферного раствора и сенсибилизатора - эозината натрия, измерении силы тока в ячейке и по достижении постоянства тока продувание воздухом раствора в ячейке, в течение 1-2 мин, облучение светом и измерение время генерации, пошедшее на восполнение убыли йода, определение количества анализируемого препарата по калибровочному графику по изменению силы тока и времени генерации йода.
Сущность заявляемого изобретения состоит в том, что в ячейке происходит изменение количества йода в результате химического взаимодействия препарата с реагентом, что приводит к изменению силы тока в ячейке. После достижения порога, при котором не происходит изменение силы тока в системе поглотительный раствор, содержащий остаток йода облучают светом и измеряют время генерации, необходимое для восполнения убыли йода в поглотительной ячейке. Количество препарата в пробе определяют по градуировочным графикам. Применение фотогенерированного йода в качестве титранта позволяет увеличить скорость окисления в результате получения атомарного йода, что приводит к уменьшению продолжительности определения.
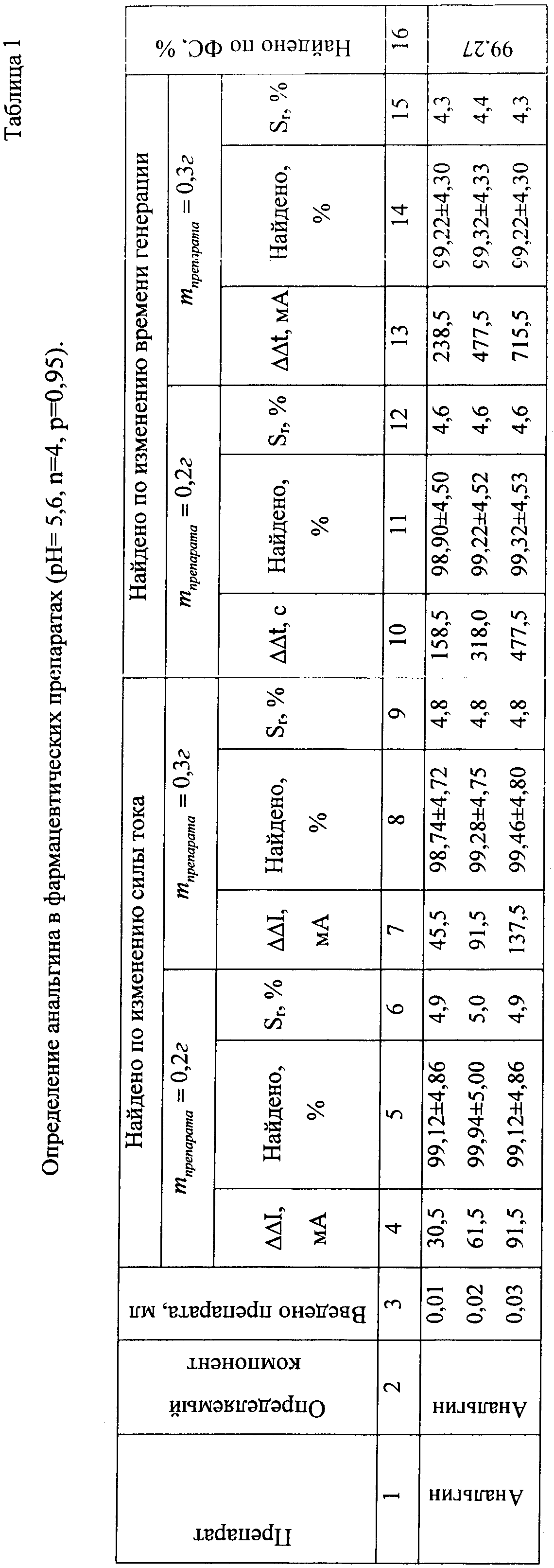
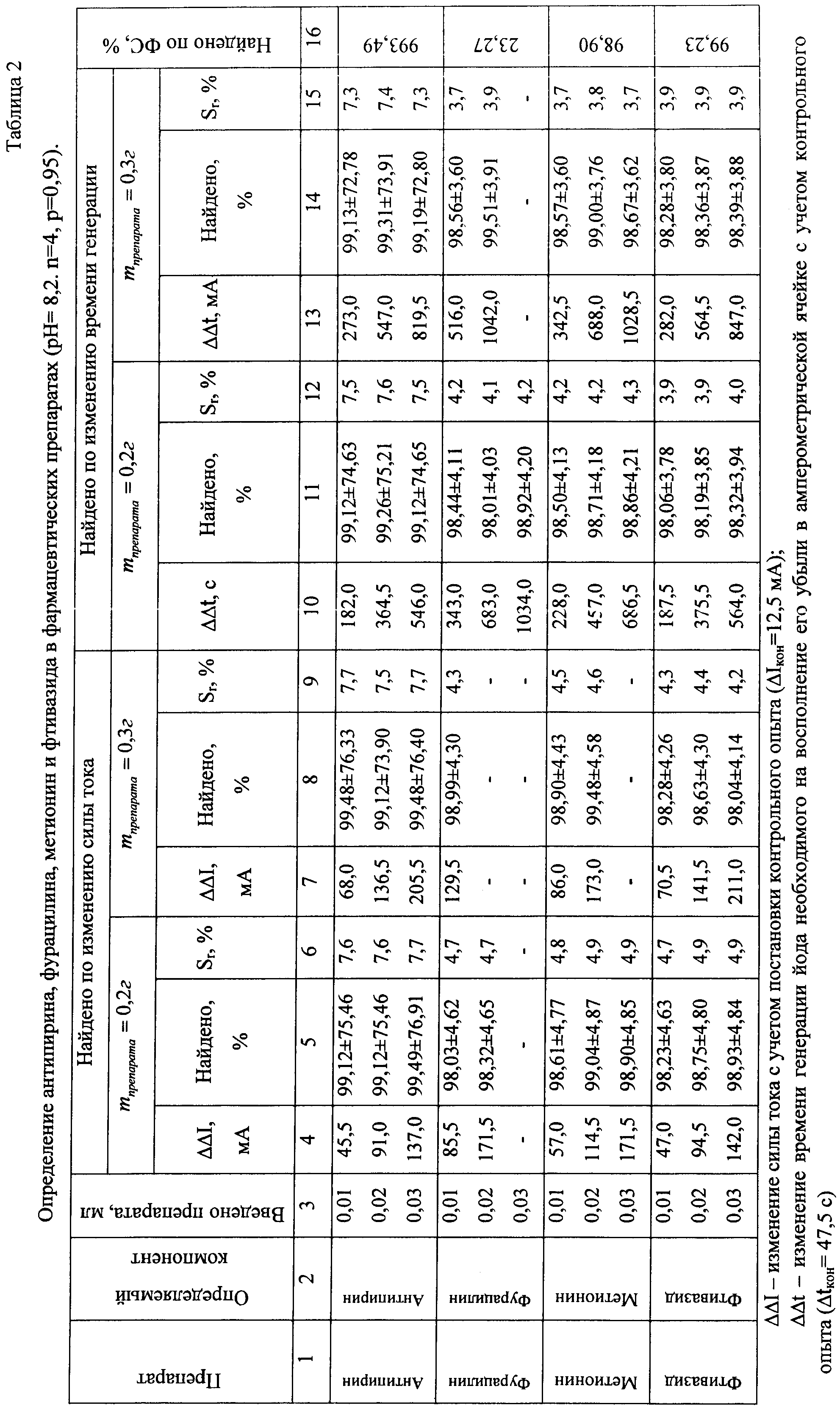
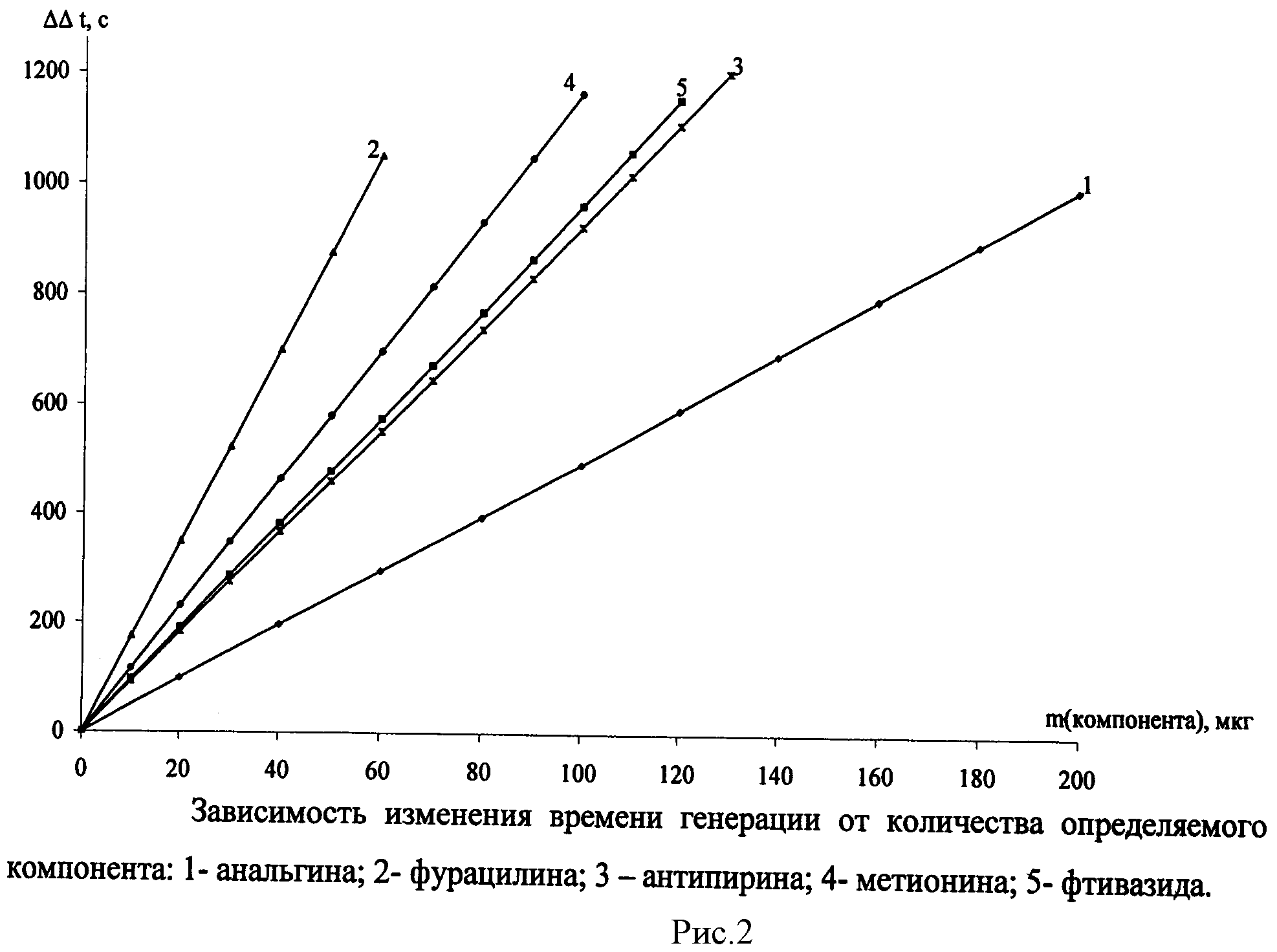
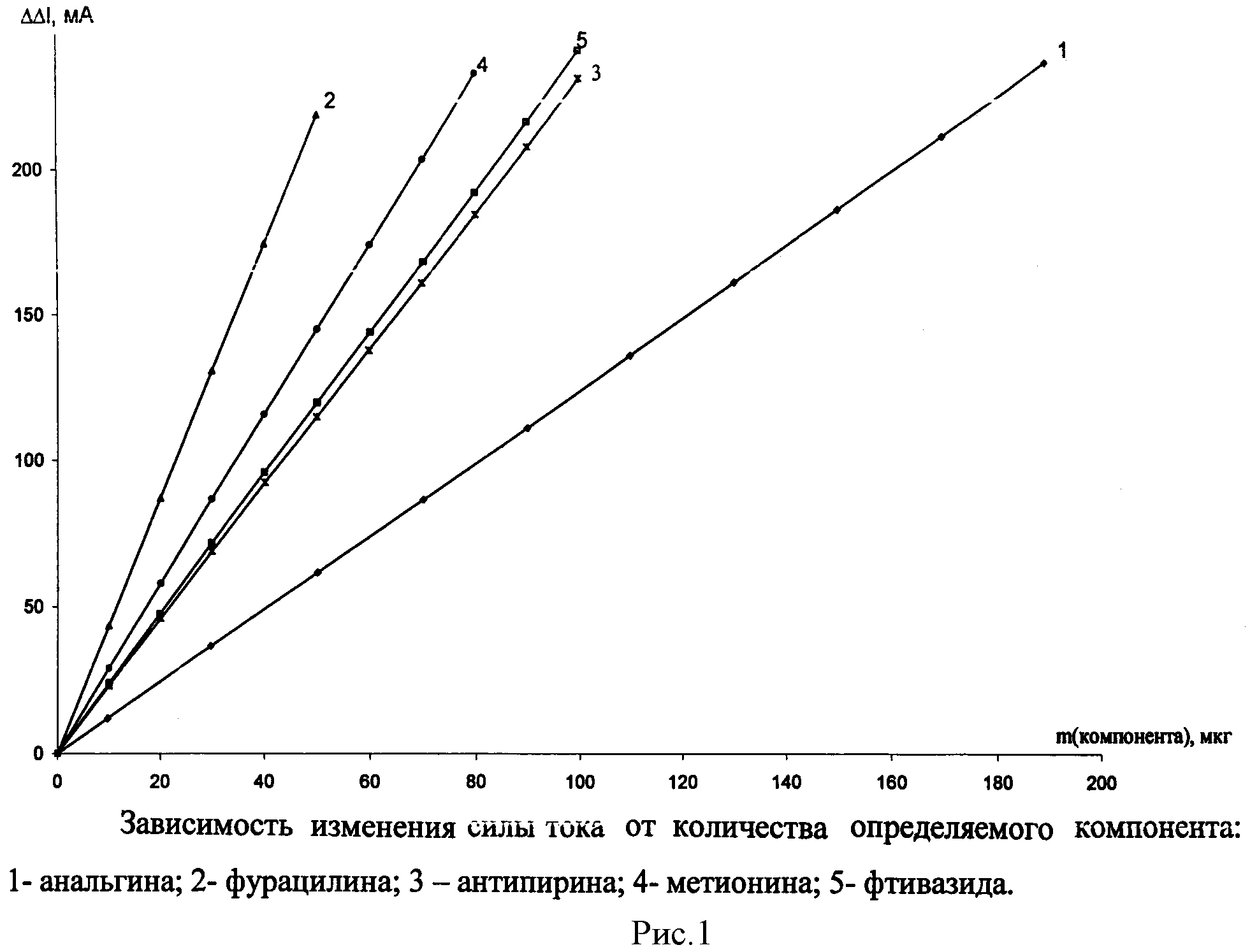
См. рис.1, 2. Результаты определения приведены в таблицах 1, 2.
Способы, рекомендованные ГОС фармацией, - классические титриметрические методы, требующие большей массы навески препарата, предварительной пробоподготовки и, как следствие, длительны в исполнении. Ошибка классических титриметрических методов достаточно высокая. Так как индикация точки эквивалентности осуществляется с помощью индикатора (крахмала) или по изменению собственной окраски титранта, точность и воспроизводимость метода малы. Предложенный метод автоматизирован, т.е. исключаются визуальная ошибка и ошибка экспериментатора, метод практически не требует предварительной пробоподготовки. Низкие затраты на реактивы, так как установка, а следовательно, и сама система могут быть использованы многократно.
Для определения достоверности полученных результатов использовались метод калибровочного графика и метод добавок.
Осуществление способа приведено в примере 1.
Пример 1. Пробоподготовку проводили по методикам, указанным в Государственной фармакопее СССР. М., 1968.
Определение анальгина (производное пиразолона-1-фенил-2,3-диметил-4-метиламинопиразолон-5-N-метансульфонат натрия). Навеску препарата массой 0,2 г и 0,3 г помещали в сухие мерные колбы объемом 100 мл, прибавляли 20 мл спирта, 5 мл 0,01н. раствора соляной кислоты. Объем в колбах доводили водой до метки.
В амперометическую ячейку помещают 40 мл 0,5 М раствора йодида калия и буферный раствор - 20 мл ацетатного буферного раствора с рН 56, 10 мл раствора эозината натрия. В раствор помещают два платиновых микроэлектрода, на которые подается разность потенциалов 0,04 В. При постоянной скорости перемешивания на раствор действуют светом от стабилизированного источника. О концентрации йода судят по изменению тока в цепи по шкале гальванометра.
После генерирования определенного количества йода отключают источник света и вводят в ячейку 0,01-1,00 мл раствора анализируемого вещества (анальгина), фиксируя при этом изменение показаний гальванометра. После достижения постоянства силы тока ячейку продувают воздухом в течение 1-2 мин, облучают светом и измеряют время генерации, пошедшее на восполнение убыли йода.
Для проведения последующих определений раствор, находящийся в амперометрической ячейке, снова облучают светом, как описано выше, генерируя в нем определенное количество йода. Одну и ту же систему используют для определений 8-12 раз. Содержание анальгина в анализируемом образце определяют по калибровочным графикам, полученным по стандартным растворам (рис.1, 2). Достоверность полученных результатов контролировали по стандартной методике и методом добавок. Результаты определения анальгина в фармацевтических препаратах приведены в таблице 1.
Пример 2. Пробоподготовку проводили по методикам, указанным в Государственной фармакопее СССР. М.: Медицина, 1968.
а) Определение антипирина (производное пиразолона-1-фенил-2,3-диметил-пиразолон-5). Навески препарата массой 0,2 г и 0,3 г помещали в мерные колбы объемом 100 мл, прибавляли 2 г ацетата натрия. Объем в колбах доводили водой до метки.
б) Определение фурацилина (производное нитрофурана-5-нитрофурфурол семикарбазон). Навески препарата массой 0,2 г и 0,3 г помещали в мерные колбы объемом 100 мл, прибавляли 10 г хлорида натрия, 50 мл дистиллированной воды и нагревали на водяной бане (70-80°С) до полного растворения. После охлаждения объем в колбах доводили водой до метки.
в) Определение фтивазида (производное изоникатиновой кислоты- изоникотиноил-(3-метокси-4-оксибензаль)-гидразон). Навески препарата массой 0,2 г и 0,3 г помещали в мерные колбы объемом 100 мл, прибавляли 2,5 мл концентрированной серной кислоты, 50 мл дистиллированной воды и кипятили в течение 2-3 минут. Растворы охлаждали и доводили водой до метки.
г) Определение метионина (аминокислота-α-амино-γ-метилтиомасляная кислота). Навески препарата массой 0,2 г и 0,3 г помещали в мерные колбы объемом 100 мл, прибавляли 0,5 г двухзамещенного фосфата калия, 0,2 г однозамещенного фосфата калия, 0,2 г йодида калия. Объем в колбах доводили водой до метки.
В амперометическую ячейку помещают 40 мл 0,5 М раствора йодида калия, буферный раствор - 20 мл 0,1 М раствора бикарбоната натрия, 10 мл раствора эозината натрия. В раствор помещают две платиновых микроэлектрода, на которые подается разность потенциалов 0,04 В. При постоянной скорости перемешивания на раствор действуют светом от стабилизированного источника. О концентрации йода судят по изменению тока в цепи по шкале гальванометра.
После генерирования определенного количества йода отключают источник света и вводят в ячейку 0,01-1,00 мл анализируемого вещества, фиксируя при этом изменение показаний гальванометра. После достижения постоянства силы тока ячейку продувают воздухом в течение 1-2 мин, облучают светом и измеряют время генерации, пошедшее на восполнение убыли йода.
Для проведения последующих определений раствор, находящийся в амперометрической ячейке, снова облучают светом, как описано выше, генерируя в нем определенное количество йода. Одну и ту же систему используют для определений 8-12 раз. Содержание анализируемых веществ определяют по калибровочным графикам, полученным по стандартным растворам (рис.1, 2). Достоверность полученных результатов контролировали по стандартной методике и методом добавок. Результаты определения компонента в фармацевтических препаратах приведены в таблице 2.
Таким образом, предложен экспрессный, высокочувствительный метод определения действующего начала производных пиразолона, нитрофурана, изоникотиновой кислоты, аминокислот в лекарственных формах.
Способ получения 4-оксоалкан-1,1,2,2-тетракарбонитрилов
Способ получения производных 4-арил-2-оксо-2н-хромен-3-карбонитрилов
Способ определения содержания аскорбиновой кислоты в растительном сырье
Способ определения микропримесей мышьяка и сурьмы в растительном лекарственном сырье
Способ получения метилового эфира 2-галоген-6-алкил-3-цианоизоникотиновых кислот
Способ изготовления индикаторной пластины для определения микропримеси свинца в растительном сырье
Способ получения 7-имино-6-оксабицикло[3.2.1]окт-3-ен-1,8,8-трикарбонитрилов
Способ получения жёсткого пенополиуретана
Лаковая композиция на основе олигоуретанакрилата
Резиновая смесь

